クマによる耐糖能研究:冬眠によるインスリン抵抗性を引きづらない方法

Saxton MW, Perry BW, Evans Hutzenbiler BD, Trojahn S, Gee A, Brown AP, et al. Serum plays an important role in reprogramming the seasonal transcriptional profile of brown bear adipocytes. iScience. 2022 Oct;25(10):105084. https://www.cell.com/iscience/fulltext/S2589-0042(22)01356-6
ハイライト
-グリズリーベアーの冬眠は、インスリン抵抗性によって特徴づけられる
-熊の脂肪細胞を、活動中の熊と冬眠中の熊の血液血清で刺激した。
-血清は、インスリンシグナルに関連する劇的な遺伝子発現反応を誘発した。
-8つの血清タンパク質がこの転写反応の駆動に関与していた
要約
細胞内で代謝の初期化がどのように行われるかを理解することは、様々な代謝異常の治療の進展に役立つと考えられる。ヒグマは、冬眠中の可逆的なインスリン抵抗性を含め、インスリン感受性の季節的なシフトを起こす。ヒグマの脂肪細胞のRNA配列解析と血清のプロテオミクスを行い、可逆的なインスリン抵抗性の原因と考えられる変化を同定した。その結果、細胞や血清の由来する季節に依存した劇的な転写の変化が観察された。脂肪細胞の遺伝子発現が大きく変化しているにもかかわらず、インスリン感受性の季節的なシフトに関連するのは8つの循環タンパク質の変化のみで、その中にはこれまでグルコースホメオスタシスと関連しなかったタンパク質も含まれていることが確認された。同定された血清タンパク質は、冬眠中の脂肪細胞を活動的な状態に移行させるのに十分である可能性がある。

熊は冬眠中にインスリン抵抗性を獲得することが判明した。しかし、血糖値とインスリンのレベルは安定しており、糖尿病になることはない。そして春になり、クマが再び外出するころには、インスリン感受性が回復している。クマの体がどのようにしてこのような快挙を成し遂げるのかを正確に理解することは、人間にとっても重要な意味を持つかもしれない。
同大学のベア・センターで飼育されているグリズリーベアから採取した血清と脂肪細胞のサンプルを分析した。あるサンプルは活動期に、またあるサンプルは冬眠期に採取された。冬眠中の2週間、研究者たちはクマに蜂蜜水を与えたが、これはクマの普段の生活を多少乱すことになった。(冬眠中のクマは、食事はしないものの、目を覚まして少しは動いているのだ)。次に研究室で、研究者たちはさまざまな細胞培養と血清サンプルを混ぜ合わせた。例えば、冬眠期の脂肪細胞と活動期の血清を混ぜて、細胞内の遺伝子活性がどのように変化するかを分析したのである。そして、冬眠が中断した時期に採取した血清を、冬眠中のクマから採取した細胞に作用させたところ、その細胞は活動期の細胞と同様の遺伝子活性の変化を示すことが判明した。
https://consumer.healthday.com/9-22-one-honey-of-a-study-well-fed-bears-give-clues-to-human-diabetes-2658230821.html
序文が勉強になる
冬眠中のクマは、循環インスリンおよびグルコースレベルが年間を通じて比較的安定しているにもかかわらず、インスリン抵抗性を示す(Rigano et al.、2017年)。冬眠中、脂肪や筋肉などの組織は、もはや血液からグルコースを取り込まず、インスリンに反応してグルコースを貯蔵しません(Palumboら、1980;Riganoら、2017)。インスリン感受性は毎年春になると回復する(McCainら, 2013; Palumboら, 1980; Riganoら, 2017)。インスリン感受性の季節的逆転は、代謝性疾患の研究のための貴重な生物学的モデルとして提案されている(Frobert et al, 2020; Martin, 2008)。我々の最近の研究では、ヒグマの原組織において、インスリンシグナル伝達経路の多くの遺伝子の発現が、非冬眠状態と冬眠状態の間で大きくシフトすることが示された(Jansen et al.,2019)。これらの遺伝子発現の変化は、正確なメカニズムはまだ不明であるが、転写の季節的な制御が、少なくとも部分的に、インスリンシグナル伝達の年次変化を支えている可能性を示唆している。
培養されたヒグマ脂肪細胞は、収穫された時期に基づいて、ヒグマの多くの生理学的特性を保持する(Jansenら、2021年;Riganoら、2017年)。例えば、冬眠中または活動期のヒグマに由来する細胞培養物は、一致する血清で培養した場合、インスリン感受性のシフトがin vivoで見られるものと一致することを示した(Riganoら、2017年)。しかし、いずれかの季節に採取した細胞を反対季節の血清で培養すると、活動期に見られるグルコース取り込み反応を再現する傾向があった(Riganoら、2017)。活動期の血清を熱処理すると、冬眠中のクマ脂肪細胞におけるグルコース取り込みの刺激作用が著しく減衰した(Rigano et al.、2017)。このことは、熱処理によって変性する1つ以上のタンパク質が、細胞培養の表現型に対する血清の効果に関与していることを示唆している。活動状態と冬眠状態の切り替えに寄与する特定の血清因子を同定することは、冬眠生理学およびインスリン感受性を調節するメカニズムの理解において重要なステップである。培養クマ脂肪細胞は、クマが毎年獲得するインスリン抵抗性状態の基礎となる細胞メカニズムおよび血清の寄与を実験的に調べる機会を提供するものである。クマは毎年大量の脂肪を蓄えるにもかかわらず2型糖尿病を発症しないことから、この疾患を防ぐためにクマ独自の細胞および/または血清因子が進化した可能性がある。
一次組織において活動期と冬眠期で観察されるグローバルな遺伝子発現の変化(Jansen et al., 2019)および培養脂肪細胞における標的遺伝子(Rigano et al., 2017)を考えると、培養脂肪細胞では一次脂肪組織を採取した時期によってかなりの遺伝子発現差があるだろうと推測された。冬眠中のクマ脂肪細胞と活動中のクマ脂肪細胞の間のこれらの遺伝子発現の違いは、細胞状態、血清成分、またはその2つの組み合わせによって駆動されている可能性がある。外来因子によって引き起こされる可能性のある大規模な季節変化を切り離すために、我々は以前、冬眠中の10日間、飼育下のヒグマにブドウ糖を与え、給餌試験の前後に前脂肪細胞と血清を採取した(Jansen et al.、2021)。本研究では、季節、細胞状態、血清タンパク質が脂肪細胞遺伝子発現に果たす役割を検討した。我々の目的は、エネルギー状態の機能として遺伝子発現の違いを駆動する根本的なプロセスを明らかにすることであった。冬眠中のクマに単一多量栄養素のグルコースを与えることで、冬眠中に採取した脂肪細胞の遺伝子発現の変化に関与する血清タンパク質を分離しようとする際に、日長、気温、脂肪率、食事などの交絡因子の影響を最小化することができた。
本研究の目的は、(1)培養脂肪細胞の遺伝子発現の変化を解析し、細胞培養における季節性の基盤となる生物学的プロセスを明らかにすること、(2)季節による血清変化の脂肪細胞遺伝子発現への影響を明らかにすること、(3)冬眠中に摂食したクマの血清が遺伝子発現、特にインスリンシグナルに関連して変化を与えるかどうかを明らかにすることであった。また、季節的に存在する循環タンパク質と冬眠中の摂食後の循環タンパク質を比較し、細胞機能の変化に関与している可能性のあるタンパク質を特定した。
予測は以下の通りである。(1)活動期と冬眠期に採取した細胞間で、ユニークな遺伝子ファミリーの発現が異なる (2)血清は季節的な脂肪細胞遺伝子発現の決定における主要な要因である (3)脂肪細胞から採取した血清で処理した細胞における遺伝子発現は、冬眠期と活動期で異なる (4)脂肪細胞は、冬眠期に採取した血清で処理した細胞で処理した細胞で異なる
Translated with DeepL
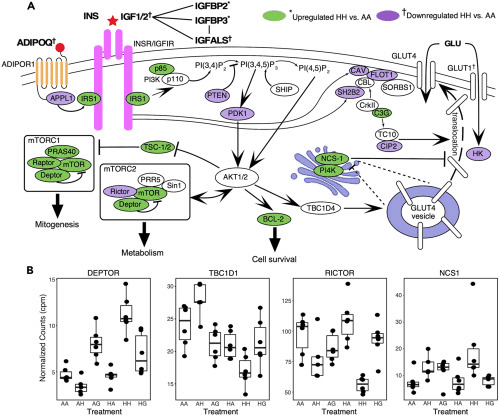
ヒグマ脂肪細胞の季節および摂食によって誘発されるインスリン感受性の上昇のいくつかの潜在的な原因を特定した。例えば、$${H_H}$$はmTORC2に特異的な遺伝子(例えば、RICTOR、PRR5L)のダウンレギュレーションを示し、それによってAKTに対する正のフィードバックを減衰させる可能性があった(Wu and Storey、2021年)。AKTの活性化は、インスリンを介したグルコースの取り込みのために、GLUT4小胞の細胞表面への移動をもたらす(Caleraら、1998年)。mTORC2複合体の他のタンパク質をコードするMTORおよびDEPTORの発現も増加したが、RICTORはしばしば、脂肪におけるインスリンの効果および全身のグルコース恒常性の仲介に主要な役割を果たす(Kumarら, 2010)。インスリンを介したグルコース取り込みの上流制御因子もまた、$${H_H}$$で差次的に発現していた。インスリンが存在するときにインスリン受容体に結合し、PI3Kの活性化を含む複数の下流イベントを誘発するタンパク質をコードするIRS1は、$${H_H}$$で発現が増加していた。ホスファチジル-3,4,5-トリフォスフェートをホスファチジル-3,4-ビフォスフェートに変換することによってインスリンによるグルコースの取り込みを阻害する重要な阻害タンパク質、PTENをコードする遺伝子は、ダウンレギュレーションされていた。インスリンシグナル伝達経路のいくつかの構成要素におけるこれらの複合的な変化は、AKT2の活性化を増加させることにつながるだろう。しかし、驚くべきことに、GLUT4の発現は影響を受けなかった。したがって、NCS1やPI4Kなど、GLUT4の細胞膜への転位に影響を与える他の二次経路が関与している可能性がある。NCS1およびPI4Kは、GLUT4の細胞膜への移動を抑制する(Mora and Pessin, 2000)。実際、$${H_H}$$では両者が有意に高いレベルで発現していた。AAと比較して$${H_H}$$では、CAV1、FLOT1、およびSH2B2の発現が減少しており、この上昇は、GLUT4の細胞膜への挿入の抑制を増強し(Fecchiら、2006)、それによってグルコースの取り込みを阻害していると思われる。このプロセスは、AKT依存的なCAVおよび関連タンパク質の細胞膜への移動に必要であると考えられるが、CAV1の発現低下により、この移動が阻害されると考えられる(図3)。これらの結果は、冬眠中のグルコース輸送とインスリンシグナルのマルチレベルの制御を示唆するものである。
GLUT4とは対照的に、GLUT1の発現は$${H_H}$$ vs AAで大きく抑制され、これは生体内で観察される効果とは逆のものであった(Jansen et al., 2019)。GLUT1は構成的なグルコース取り込みトランスポーターであるため、上記のようにインスリン経路の他のメンバーが抑制されると、インスリン存在下でも培養細胞でのグルコース取り込みが抑制されることは確かにもっともである。GLUT1発現の抑制はHA細胞では完全に逆転したが、HG細胞では逆転しなかったことから、いくつかのグルコース取り込み経路が細胞自律的に制御されていることが示唆される。このことを裏付けるように、$${H_H}$$細胞ではHKの発現も抑制されたが、HG細胞では抑制されなかった。これは、以前に報告された解糖の一般的な減少(Jansenら、2019)およびin vitroで観察された好気性呼吸(Hoganら、2022)と一致するだろう。