
ERJ誌:喘息における気道リモデリングと上皮:新たな時代の幕開けに

まずスペルについて
Claudeによると・・・
Both "remodeling" and "remodelling" are correct spellings, but they differ in their usage based on the variety of English:
- Remodeling is the preferred spelling in American English. It refers to the renovation or reconstruction of part or all of a building or structure.
Examples:
"We are planning a kitchen remodeling project."
"The house underwent a complete remodeling after being damaged in the storm."
- Remodelling is the preferred spelling in British English and other varieties that follow British conventions. It has the same meaning as the American "remodeling."
Examples:
"They hired contractors for a bathroom remodelling."
"The office space required extensive remodelling to meet the new safety codes."
So in summary, the spelling with one 'l' (remodeling) is standard in American English, while the spelling with two 'l's (remodelling) is standard in British English. Both are correct in their respective dialects when referring to construction or renovation work.
米国ジャーナルに慣れてると違和感を感じてしまう
で、以下、ERJだから英国英語にてremodellingを使用
ChatGPT4にて主に要約・翻訳使用
Varricchi, Gilda, Christopher E. Brightling, Christopher Grainge, Bart N. LambrechtとPascal Chanez. 「Airway remodelling in asthma and the epithelium: on the edge of a new era」. European Respiratory Journal 63, no. 4 (2024年4月): 2301619. https://doi.org/10.1183/13993003.01619-2023 .
喘息は気道の慢性的で異質な疾患であり、通常、気道リモデリングとして知られる構造変化によって特徴づけられます。環境的な侮辱、包括的には病原体、アレルゲン、汚染物質に対する反応として、上皮細胞は構造および免疫細胞の両方に下流効果を持つ多種多様な媒介物質を含む炎症カスケードを介してリモデリングを開始することができます。
これらの媒介物質には、上皮細胞のサイトカインである胸腺間質リンパポエチン、インターロイキン(IL)-33、およびIL-25が含まれ、これらは上皮細胞と線維芽細胞、肥満細胞と気道平滑筋細胞との間のクロストーク、さらにマクロファージなどの免疫細胞とのシグナリングを通じて気道リモデリングを促進します。上皮細胞は、気管支収縮中に存在する機械的ストレスに対する反応として、炎症とは無関係に気道リモデリングを開始することもできます。
さらに、上皮成分への遺伝的およびエピジェネティックな変更がリモデリングに影響を与えると考えられています。
ここでは、遺伝子シークエンシングやイメージング技術の発展によって促進される、上皮および上皮サイトカインが気道リモデリングを推進する役割の理解に関する最近の進歩を概説。また、上皮および上皮サイトカインを標的とする新規および既存の治療薬が気道リモデリングをどのように変更できるかについても探求。
序文要約
喘息は気道の慢性的な炎症性疾患であり、喘鳴、胸部の締め付け、咳、呼吸困難、呼気流量制限が変動する症状が特徴です。
疾患は表現型が異質で、発症年齢、重症度、治療への反応が患者ごとに異なります。
生物学的および免疫学的メカニズムの異質性は、細胞レベルと分子レベルで類似した疾患特性を持つ患者グループを定義する「エンドタイプ」の概念を導入しました。
最も一般的な炎症性エンドタイプは、T2(タイプ2)ハイ喘息で、高レベルのT2炎症バイオマーカーを示します。
T2 low 喘息の患者はT2炎症が低く、優勢な炎症細胞は好中球であるか、または炎症細胞が極端に少ないことがあります。
エンドタイプと表現型は動的であり、異なる環境刺激が生涯を通じて表現型を変化させる可能性があります。
気道上皮は喘息の病理生理において重要な役割を果たし、外部環境からの構造的および免疫学的なバリアとして機能します。
空気中の病原体や汚染物質、アレルゲンにより上皮細胞が損傷し、炎症を引き起こす上皮サイトカインが放出されます。
喘息患者では、異常な免疫反応と修復過程が慢性炎症と上皮損傷を引き起こし、気道リモデリングとして知られる構造変化が生じます。
気道リモデリングは喘息のほぼ普遍的な特徴であり、病状の重症度が増すにつれて悪化し、疾患の慢性化や機能低下を招きます。
気道リモデリングはかつて慢性炎症の結果として後期に発生する二次現象と考えられていましたが、若年児の生検研究はそれが喘息の早期発生事象であることを示しています。
遺伝子シークエンシングとイメージング技術の発展は、上皮と上皮サイトカインが気道リモデリングを推進する役割の理解を深め、新しい治療薬の開発の可能性を広げています。
How the epithelium orchestrates airway remodelling:上皮が気道リモデリングをどう指揮する?
気道の上皮細胞は喘息における気道リモデリングの開始点として機能します。
上皮細胞は、病原体、アレルゲン、環境的侮辱(汚染物質や毒素など)による細胞損傷から派生する病原体関連分子パターンおよび損傷関連分子パターンを検出するパターン認識受容体を発現します。
パターン認識受容体の活性化により、上皮サイトカインおよびILを含むケモカインとサイトカインが放出され、様々な免疫細胞と構造細胞を巻き込む下流の炎症カスケードが引き起こされます。
プロ炎症性刺激とサイトカインはまた、誘導型一酸化窒素合成酵素の発現を引き起こし、一酸化窒素レベルの増加をもたらします。これにより、増殖、アポトーシス、移動に関与するタンパク質の硝酸化を含む慢性炎症反応が誘発されます。
環境的侮辱は、上皮細胞のアポトーシスを誘発し、TGF-βなどの媒介物質の放出を伴います。これは組織再生プロセスの開始を試み、恒常性の回復を促します。
気道の壁が折りたたまれる際に、圧縮(細胞が押し合う)、伸展(上皮折り目の頂点の細胞が伸びる)、せん断応力(気道の直径の減少とともに空気の速度が増加する)などの機械的力が上皮に影響を与えます。
実験モデルを用いて、機械的力が炎症とは無関係に気道リモデリングを引き起こすことが示されています。喘息患者が経験する気管支収縮中に、上皮細胞は過度の機械的力にさらされます。
機械的ストレスは、炎症がない状態でもリモデリングを促進する反応を引き起こし、フィブロネクチンとコラーゲンIIIおよびVの生産の増加を通じて粘膜下の肥厚を引き起こします。
気道リモデリングは自己増殖的な性質を持ちます。上皮の破壊は異常な上皮媒介免疫応答を引き起こし、気道内の細菌や真菌などの微生物の持続を促進する可能性があります。
Role of epithelial cytokines in airway remodelling:気道リモデリングにおける上皮サイトカインの役割
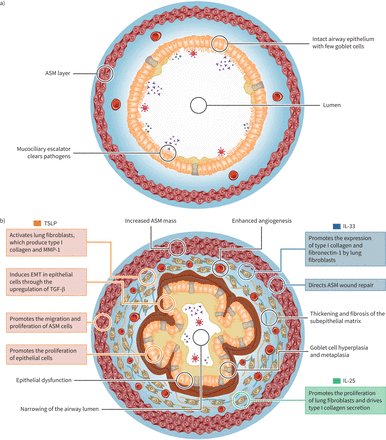
胸腺間質リンパポエチン(TSLP)、IL-33、IL-25という三つの上皮サイトカインは、「アラーミン」として知られ、喘息の病理生理に中心的な役割を果たしています。これらは、T2型および非T2型炎症、さらに気道の構造変化を含む先天的および適応的免疫応答を仲介する主要な調節因子として機能します。
喘息患者において、TSLPとIL-33の気道レベルは健康な個人と比較して高く、病気の重症度と相関しています。IL-25の痰中濃度も病気の重症度と関連があるかもしれません。
これらの上皮サイトカインは、肺線維芽細胞や気道平滑筋(ASM)細胞などの間葉細胞に多様でしばしば重なる影響を及ぼします。TSLPは肺線維芽細胞を活性化し、コラーゲンIやMMP-1などの細胞外マトリックス(ECM)分子の産生を促進します。
IL-33は肺線維芽細胞においてコラーゲンとフィブロネクチン-1の発現を促進し、IL-25は肺線維芽細胞の増殖とこれらの細胞によるコラーゲンの分泌を促進します。
上皮から間葉細胞への遷移(EMT)の間、上皮細胞は上皮マーカーを失い、基底膜へ移動して間葉マーカーを獲得し、ECMを合成します。これは傷の治癒中に損傷した上皮を基底細胞が置き換えるための枠組みを提供します。
TSLPとIL-33は気道平滑筋(ASM)と肥満細胞の間の相互作用を仲介し、気道構造と機能に影響を与えます。TSLPはASM細胞の増殖と移動を促進し、これらの細胞からの炎症性サイトカインの放出を促進します。
肥満細胞は喘息の病理生理および気道リモデリングにおいて重要な役割を果たし、多くのサイトカイン(例:TSLP, IL-33, IL-25, IL-4, IL-13, TGF-β)や血管新生因子(例:VEGF-A)、その他の炎症媒介物質や気管支収縮剤の放出に関与します。
IL-25はマウスの喘息モデルにおいて、気道の血管新生を誘導することにより気道リモデリングに寄与します。
Downstream immune cell actions influence airway remodelling:下流免疫細胞活動は気道リモデリングへ影響

EGFR:表皮成長因子受容体;IL:インターロイキン;ILC2:第2群固有リンパ細胞;R:受容体;RAGE:終末糖化産物受容体;ST2:血清刺激-2;TSLP:胸腺間質リンパポエチン。
T2型炎症応答中、TSLPは樹状細胞を活性化し、これがさらにナイーブなヘルパーT細胞を刺激してIL-4、IL-5、IL-13などのTh2様サイトカインを産生させます。
IL-33はTSLPと樹状細胞のシグナリングのポジティブな調節因子として機能し、Th2細胞による炎症応答の開始と維持を促進します。
TSLP、IL-33、IL-25はILC2s(インナクティブリンパイド細胞2型)を活性化し、これらもIL-4、IL-5、IL-13を産生します。
Th2細胞とILC2sから分泌されたIL-5は、好酸球の募集、成熟、生存に影響を与えることで好酸球性炎症を刺激します。
活性化された好酸球と肥満細胞はシステイン酸ロイコトリエンを放出し、これらは強力な気管支収縮物質であり、ASM細胞の増殖とTGF-β、カチオン性タンパク質、サイトカインの放出を通じて気道リモデリングを引き起こします。
MET遺伝子(肝細胞増殖因子受容体をコード)とMMP10遺伝子(MMP-10をコード)は、粘膜下好酸球と関連する気道リモデリングと細胞炎症に関与しています。
IL-4とIL-13は、杯細胞の増殖を介した粘液生成とコラーゲン沈着を通じて粘膜下線維症を強化します。
TSLPはT2炎症非依存的な気道リモデリングにも役割を果たすかもしれません。TSLPによって活性化された樹状細胞はナイーブなT細胞をTh17型に極性化させることができます。
樹状細胞とTh17サイトカインの協働作用は好中球性炎症とTGF-βの発現と相関する線維化マトリックス成分の蓄積を促進します。
これら3つの上皮サイトカインは人間の肺マクロファージを活性化することができます。マクロファージは貪食、サイトカインおよび血管新生因子の産生、肺の組織修復の調節を通じて気道の炎症反応に影響を与えます。
肺のマクロファージの2つの異なる集団が特定されており、一方は主に神経に隣接して抗原を提示し、もう一方は血管のそばに存在し、マウスモデルで炎症と線維症を抑制する重要な役割を持つことが示されています。
Influence of genetic and epigenetic changes in the airway epithelium on remodelling:リモデリングにおける気道上皮における遺伝・エピジェネティックの影響
気道上皮の遺伝的変異は、喘息の発症または悪化を引き起こし、リモデリングに影響を与えることがあります。
Shrineらによる中等度から重度の喘息に対する最大規模の全ゲノム関連研究では、MUC5AC、GATA3、KIAA1109の変異が中等度から重度の喘息の感受性を増加させることが報告されています。MUC5AC遺伝子の変異は粘液栓を増加させ、GATA3はT2免疫とアレルギーを調節する転写因子です。
Heijinkらは、上皮機能に関連する12個の遺伝子を選択しました。これには、炎症環境(IL33, TSLP, IL1RL1)、病原体への応答(CDHR3)、粘液線毛クリアランス(MUC5AC, KIF3A, EFHC1)、細胞ホメオスタシスと上皮の整合性に関連する遺伝子(PCDH1, SMAD3, GSDMB, ORMDL3, PLAUR)が含まれます。
Gasdermin B(GSDMB)は喘息の上皮で高発現し、遺伝子改変マウスでのGSDMBの増加発現は、気道炎症のない状態で自発的なリモデリングとAHRを引き起こします。
Orosomucoid-like protein isoform 3(ORMDL3)は上皮細胞で発現し、小胞体ストレスとスフィンゴ脂質ホメオスタシスを調節します。ORMDL3を過剰発現する遺伝子改変マウスは、気道平滑筋の質量増加、粘膜下線維症、粘液の増加を示します。
Plasminogen activator urokinase receptor(PLAUR)は上皮で発現し、ウロキナーゼ型プラスミノーゲンアクチベーターの活性化を調節し、上皮の修復、増殖、リモデリングに関与します。喘息患者では、PLAURの変異は肺機能の急速な低下と基底膜の厚さ、コラーゲンIIIの沈着、基底上皮の増殖と関連する気道リモデリングと関連しています。
YKL-40は気道上皮で発現するキチン結合グリコタンパクで、その血清レベルの上昇は喘息の重症度、気道リモデリング、RBMの厚みの増加と相関します。
上皮細胞のDNAメチル化パターンは、健康な子供やアトピー性喘息の子供と比べて、喘息の子供で異なります。学齢期の子供では、上皮バリア機能、気道上皮の整合性、免疫調節に関連する遺伝子のメチル化が異なります。
遺伝子の発現の過程での炎症、上皮間葉転換(EMT)、細胞外マトリックス(ECM)のリモデリングを引き起こすDNAの構造変化が起こることがあります。誘導可能なエピジェネティックなグアニン酸化再プログラミング経路を標的とすることで、前臨床モデルで気道炎症が減少しました。
Structural and clinical consequences of airway remodelling:気道リモデリングの構造と臨床的consequence
気道リモデリングに伴う超微細構造の変化により、気道壁の肥厚や腔の狭窄などの気道幾何学的変形が生じ、粘液栓や気道閉塞などの換気欠陥が発生します。これらの現象の一部は、CTなどの画像技術を使用して評価できます。
杯細胞の過形成と粘膜下腺の肥大により、痰の産生が増加し、痰分泌による気道狭窄と気道壁の厚みが増加します。これらの変化は最終的に粘液栓の形成につながり、これは重度の気流制限や死亡と関連があります。
上皮下線維症はECMの増加によって発生し、気道壁の肥厚を引き起こし、これは喘息の重症度および気道過敏性(AHR)と相関します。
増加した気道平滑筋(ASM)の質量は喘息の重症度と関連しており、ASM細胞が上皮に向かって移動し、その後のASM内のECMの増加が気流閉塞に寄与する可能性があります。
炎症性血管新生による気道壁微小血管の変化が気道壁の浮腫を引き起こし、これが腔の狭窄に寄与します。
気道の軟骨量の減少と軟骨プロテオグリカンの分解の増加が慢性気道閉塞に寄与し、与えられたASM収縮度に対してより強力な気管支収縮を可能にします。
胸部レントゲン写真やCTスキャンの質的評価は喘息の標準治療の一部ですが、他の肺画像プラットフォームやソフトウェアアルゴリズムは、地域の気道構造と機能、および気道炎症を定量化できます。CT画像に加えて、超分極ガスMRIやシングルフォトン放出CTなどの新しいアプローチが重症喘息の異質性を理解するのに役立ち、精密医療に向けた臨床管理の進展が期待されます。
気道の幾何学を測定することで、個々の気道レベルでのリモデリングの影響を測定し、肺機能試験は臓器レベルでのリモデリングを測定します。しかし、リモデリングにつながる超構造的/細胞的変化は、臨床症状(例えば息切れ)の発展と同じ時間枠で起こりません。異なるモダリティからの情報へのアクセスと組み合わせることで、気道リモデリングの基本的なメカニズムについての洞察が得られます。
Identification of airway remodelling phenotypes in asthma:喘息における気道リモデリングphenotypeの同定
気管支生検および死後組織切片研究は、異なる喘息表現型の患者間で気道の構造的特徴に違いがあることを示しています。
重度の喘息患者で好酸球性炎症が高い場合、好酸球がほぼ存在しない患者よりも基底膜(RBM)の厚みが大きいです。
初発が幼少期の喘息患者と成人発症の喘息患者とでは、RBMの厚さに違いは見られませんでしたが、成人発症の患者は肺機能(FEV1や強制肺活量)が劣り、病気の期間が短いです。
成人発症の喘息患者のうち、好酸球性炎症がある者は、炎症がない者よりもRBMの厚化が大きいです。
少顆粒細胞型喘息の患者は、健康な個体と比較して、気道平滑筋(ASM)層とRBMの厚みが増加しています。
顆粒球性喘息の患者も気道壁の厚みが増加し、ASMの短縮と粘液の閉塞により気道の径が狭くなっています。
定量的CTイメージングを使用して気道リモデリングの表現型を特定しようとする試みも行われています。
大気道の壁と腔の体積に関するクラスター分析により、すべて気道閉塞を示す3つの表現型が特定されました:1つは気道壁と腔の体積が増加し、もう1つは腔の狭窄があり、どちらも肺機能が低下しているクラスターと、気道リモデリングがなく臨床的に軽度の疾患を持つ第3のクラスター。
重度喘息患者のコホートにおいて、大気道と小気道を通じたリモデリングの2回目の分析では、3つの表現型が同定されました:1つは大から中程度の気管支壁の肥厚、粘液栓と気管支拡張を伴い、全身的な好酸球性炎症と関連しています;もう1つは小気道リモデリングと固定気流閉塞を特徴とし、男性、喫煙者、コントローラー薬の使用が多いことと関連しています;最後の1つは好酸球性炎症も気道リモデリングもないものです。
リモデリング表現型の正確な同定は、気道リモデリングに特化した特性を治療するための標的アプローチの開発を可能にするかもしれません。これにより、肺機能の低下を防ぐことが目標です。患者の臨床、炎症、リモデリングの表現型の組み合わせが、その治療に最適な個別化されたアプローチを決定することになります。
Targeting the epithelium and epithelial cytokines to modify airway remodelling:上皮および上皮サイトカインの気道リモデリングへのtargeting
喘息の様々な治療法は、直接的または間接的に上皮を標的とし、臨床的な症状の改善だけでなく、FEV1やその他の肺機能試験パラメータによって測定される気道リモデリングや肺機能の低下の特徴を改善することもあります。
Glucocorticoids
喘息の一次治療には通常、上皮細胞を含むほぼ普遍的に発現するグルココルチコイド受容体を標的とした吸入ステロイド(ICS)が含まれます。ICSの維持使用(1年以上)は、FEV1(一秒量)の適度な改善と関連しています。
グルココルチコイドは慢性炎症を軽減し、これが間接的に気道リモデリングに寄与します。しかし、ICSまたは経口ステロイドが気道リモデリングに直接有益な影響を与えるという証拠は矛盾しています。高用量のICSは、軽度から中等度の喘息患者において粘膜下血管と基底膜の厚さを減少させました。グルココルチコイド治療は、タイトジャンクションタンパク質の再配布を通じて上皮細胞単層の完全性を回復させることも示されています。さらに、上皮細胞での研究により、グルココルチコイド暴露が杯細胞過形成を減少させる可能性が示唆されていますが、軽度の喘息患者でのICS治療ではこの点が確認されていません。
実際には、グルココルチコイド治療がカスパーゼ媒介の上皮細胞アポトーシスを誘発することにより、気道リモデリングに寄与する可能性があります。
Biologics targeting T2 inflammation:T2炎症へのbilogicsのtargeting
バイオロジック治療は中等度から重度の喘息患者の病状コントロール向上のために、追加維持治療として処方されることがあります。
モノクローナル抗体(mAbs)はIgEやTh2サイトカイン(IL-5、IL-4、IL-13)およびその受容体を標的とし、これらの免疫細胞媒介物質に対する作用を通じて上皮に間接的な影響を与えます。
Omalizumabは自由なIgEに結合し、そのマスト細胞、好塩基球、樹状細胞上の高親和性IgE受容体FcεRIへの結合を阻害します。RCTではFEV1(一秒量)の改善は見られませんでしたが、実世界データではFEV1の改善と重症増悪の減少が示されています。
MepolizumabとreslizumabはIL-5に結合し、それが好酸球上のIL-5受容体αサブユニットに結合するのを防ぎます。これらのバイオロジックは重症好酸球性喘息患者でのRCTおよび実世界設定でFEV1を改善しました。
Dupilumabはリンパ球および上皮細胞上に存在するIL-4受容体αサブユニットに結合し、IL-4とIL-13のシグナリングをブロックします。これにより、粘液産生、杯細胞過形成、粘膜下線維症、コラーゲン沈着が減少する可能性があります。重症喘息患者では、RCTおよび実世界研究でFEV1と他の肺機能指標が改善しました。
IL-13に対する抗体であるlebrikizumabは承認されていない治療法ですが、喘息におけるIL-13の役割をさらに示す証拠として、未コントロール喘息患者の粘膜下コラーゲンの厚みを減少させました。
IL-4とIL-13に対するワクチンの組み合わせ接種は、喘息のマウスモデルで予防および治療効果を示し、治療抗体に対する費用効果の高い代替手段としての臨床開発の道を開きました。
Biologics targeting epithelial cytokines:上皮サイトカインへのbiologics targeting
上皮サイトカインであるTSLP、IL-33、IL-25を抑制することで直接的に上皮を標的とすることは、気道リモデリングの改善に有望なアプローチかもしれません。
Tezepelumab(TSLPを標的とするヒトのモノクローナル抗体)は、重症喘息患者の治療に最近承認されました。フェノタイプやバイオマーカーの制限はありません。
Tezepelumabは重症でコントロールされていない喘息患者において、プラセボと比較してFEV1(一秒量)や他の肺機能指標の迅速かつ持続的な改善を示しました。
Tezepelumabは血清中のMMP-3およびMMP-10のレベルも低下させました。
Tezepelumabは、基礎となる血中好酸球数に関わらず、気管支生検における粘膜下好酸球数を有意に減少させました。
Tezepelumabは、気道のルーメン領域の拡大も示し、これは痰栓の減少と関連しており、FEV1と好酸球性炎症の改善と相関していました。
IL-33シグナリングを標的とするいくつかのバイオロジック治療法が開発中または最近開発されています。
Itepekimab(抗IL-33モノクローナル抗体)は、中等度から重度の喘息患者でプラセボに比べて12週間の治療後にFEV1を改善しました。
Astegolimab(IL-33受容体ST2を阻害するヒトモノクローナル抗体)は、重症喘息患者で54週間の治療後にFEV1を有意に改善しませんでしたが、増悪は減少しました。
Tozorakimab(IL-33活性をST2およびRAGE-EGFR複合体シグナリング経路の両方で阻害する新しい二重作用モノクローナル抗体)は、炎症バイオマーカーを低下させることが示され、現在、第2相試験が進行中です。
IL-25またはその受容体を標的とするバイオロジックは承認されておらず、後期開発段階にあるものも知られていませんが、アレルゲン誘発気道リモデリングのマウスモデルでIL-25のブロックは気道の好酸球、Th2サイトカインのレベルを減少させ、周気管支のコラーゲン沈着、気道平滑筋の過形成、気道過敏性を阻害しました。