特発性肺線維症の筋線維芽細胞の脱分化

致死性で進行性の不可逆的な疾患である特発性肺線維症の筋線維芽細胞の脱分化に焦点を当て、有望な治療アプローチとしての可能性を探っています。このレビューでは、肺線維症の発生率、治療の課題、および筋線維芽細胞の分化と脱分化におけるTGF-βシグナル伝達の役割について概説しています。さらに、Nrf2、Ca2 +、HIF-1α、BETタンパク質、MSC細胞外小胞、PPAR、MEK / ERK経路、およびcAMP / PKA経路などの筋線維芽細胞の脱分化に関与するその他の重要なシグナル伝達経路についても掘り下げています。さらに、BTZ、MyoD、SET8、および機械的力の調節などの潜在的な治療標的と薬剤を強調し、これらの発見の臨床的意義と、肺線維症の治療のための筋線維芽細胞の脱分化を標的とする将来の研究の方向性について議論しています。
Ju, Xuetao, Kai Wang, Congjian Wang, Chenxi Zeng, Yi WangとJun Yu. 「Regulation of Myofibroblast Dedifferentiation in Pulmonary Fibrosis」. Respiratory Research 25, no. 1 (2024年7月18日): 284. https://doi.org/10.1186/s12931-024-02898-9.
Myofibroblast
【筋線維芽細胞とは】
- 筋線維芽細胞は、ミオシンとアクチンなどの筋肉タンパク質を含む線維芽細胞様の細胞。
- 線維芽細胞と平滑筋細胞の中間に位置する細胞として機能。
- 傷の治癒部位や肺、腎臓、肝臓、皮膚などのリモデリング能力が高い線維化臓器に広く存在。
- 主な効果細胞として、多量の細胞外マトリックス(ECM)タンパク質(I型コラーゲンやフィブロネクチン)を生成し、傷の治癒と臓器の線維化反応を促進。
- α-平滑筋アクチン(α-SMA)は、筋線維芽細胞を線維芽細胞や他の前駆細胞と区別するための最も一般的なバイオマーカー。
- 他の潜在的マーカーには、ビメンチン、線維芽細胞特異的タンパク質1(FSP-1)、カドヘリン-11、アンジオテンシン1受容体(AT1)、TGF-βタイプII受容体(TβRII)、パキシリン、テンシン、フィブロネクチンエクストラドミナントAスプライスバリアント、フリズルド-2、オステオポンチン、テネイシンC、ペリオスチンなど。
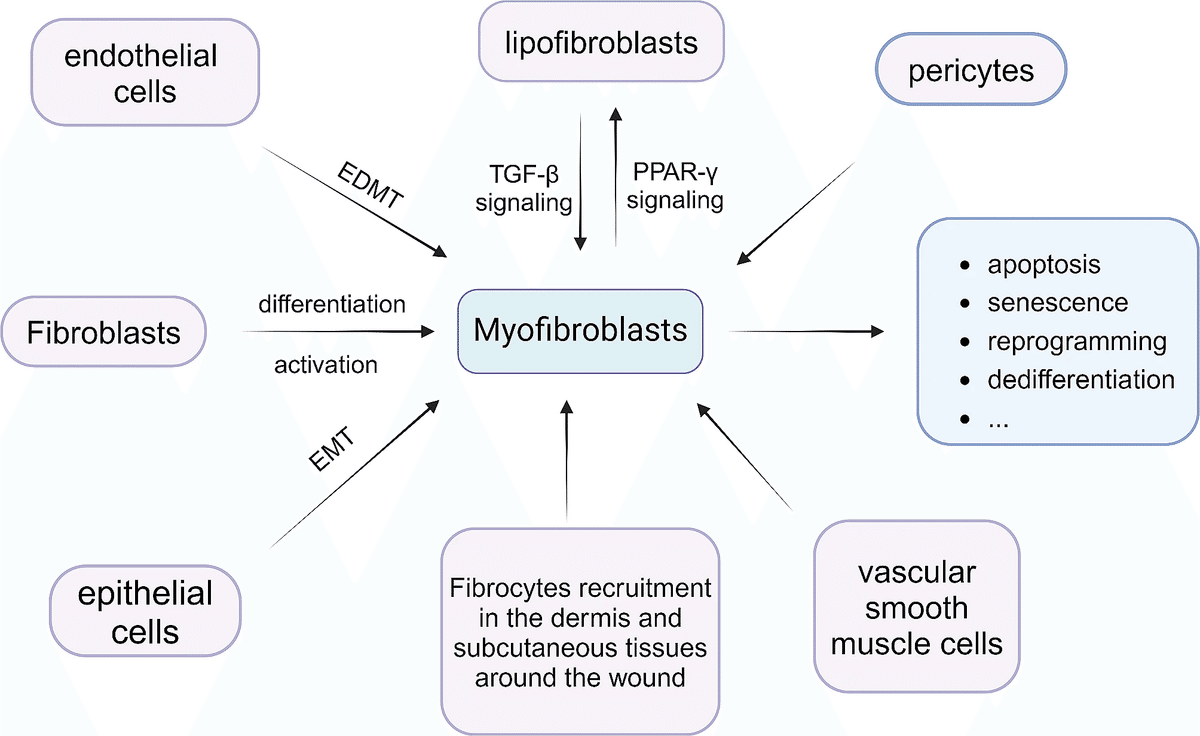
【筋線芽細胞の起源】
- 筋線維芽細胞にはいくつかの共通の起源がある。
- 線維芽細胞がTGF-β/SMAD経路や血小板由来成長因子(PDGF)によって筋線維芽細胞に分化。
- 上皮間葉転換(EMT)や内皮間葉転換(EDMT)。
- 傷周辺の真皮や皮下組織からの線維細胞の動員。
- 血管周囲細胞や血管平滑筋細胞の筋線維芽細胞への変換。
- 脂肪生成性と筋生成性線維芽細胞表現型間の相互変換。
【筋線維芽細胞の脱分化とその治療的意味】
- 肺線維症の治療には、線維化の根本原因の除去、線維性ECMの分解および除去、線維芽細胞の増殖および筋線維芽細胞への分化の抑制、筋線維芽細胞の除去(アポトーシス、老化、再プログラム化、脱分化などのプロセスの誘導)などの戦略がある。
- 脱分化は、成熟した細胞が未分化の細胞表現型に戻るプロセス。
- 筋線維芽細胞の脱分化は、ECM産生の大幅な減少、α-SMAの発現低下、およびストレスファイバーとの結合親和性の低下を指す。
- 脱分化の研究は比較的新しい分野であり、統一された研究方法がないため、異なる研究で異なる実験手法が用いられる。
- 一般的な実験手法は、DMEMで24時間の血清飢餓後、TGF-βで24~48時間の前処理を行い、線維芽細胞を筋線維芽細胞に分化させ、その後脱分化因子を適用してα-SMAおよびコラーゲンの分子発現レベルを評価する。
- 脱分化の指標として、α-SMAおよびコラーゲンの発現の大幅な減少が使用される。
【治療の可能性】
- 他の治療法は通常、肺線維症の進行を抑制し、症状を軽減し、生活の質を向上させるだけ。
- 線維化が診断される頃には、組織損傷が進行しているため、筋線維芽細胞の形成を抑制し、線維化の進行を阻止するだけでは治癒効果を得られない可能性がある。
- 筋線維芽細胞の脱分化を誘導することは、既存の肺線維症を逆転させ、治癒効果を達成する可能性があると考えられているため、筋線芽細胞の脱分化に関する研究が増加している。
TGF-β signaling

TGF-βシグナルと線維芽細胞の分化
TGF-βファミリー:
TGF-βはTGF-βファミリーのプロトタイプで、アクチビン、ノーダル、骨形成タンパク質(BMPs)、成長分化因子(GDFs)などを含む。
シグナル伝達経路:
線維芽細胞から筋線維芽細胞への分化過程で、TGF-βはTGF-β受容体II(TβRII)に認識され、TGF-β受容体I(TβRI)をリン酸化。
活性化されたTβRIはR-Smad(Smad2およびSmad3)タンパク質をリン酸化し、Co-Smad(Smad4)と三量体複合体を形成して核内に移行し、フィブロネクチン、コラーゲン、PAI-1、CTGFなどの線維化関連遺伝子の発現を調節。
共活性化因子と共抑制因子:
p300、CBP、AP-1、Sp1などの共活性化因子や、c-Ski、SnoN、Smad核相互作用タンパク質-1などの共抑制因子がこの過程を調整。
EMTとSmad経路:
Smadシグナル経路の活性化は、E-cadherinの阻害因子であるSNAIL、SLUG、ZEB、TWISTなどの転写因子の発現を誘導し、デスモソーム解離を介して上皮間葉転換(EMT)を媒介。
また、Smad経路の活性化はAktの活性化を導き、β-カテニンの核内移行を促進し、α-SMAのアップレギュレーションを引き起こす。
ネガティブレギュレーター:
Smad6およびSmad7は、タイプI受容体に結合し、Co-Smad4への結合を競合することでTGF-βシグナルを抑制。
Smad6とSmad7は、Smurf1およびSmurf2をリクルートし、Smadタンパク質をプロテアソーム分解に標的化。
TGF-βシグナルと筋線維芽細胞の脱分化
TGF-β経路の役割:
TGF-βシグナル経路は、線維芽細胞の分化および筋線維芽細胞の脱分化の調節に重要。
特発性肺線維症(IPF)の治療研究は、TGF-β経路の阻害に焦点を当て、Smad2/3のリン酸化を抑制し、Smad7を活性化することに関心。
Nrf2活性化剤:
亜ヒ酸トリオキシド(ATO)やスルフォラファン(SFN)などのNrf2活性化剤は、Smad2/3のリン酸化を抑制し、TGF-βによるROSの蓄積を減少させ、抗酸化防御を回復。
Ca2+調節:
ストア作動性カルシウム流入(SOCE)阻害剤は、TGF-β1/Smad3経路を抑制することで線維化マーカーの発現を抑制。
KCa3.1阻害剤は、Smad2/3の核内移行を妨げ、筋線維芽細胞の脱分化を誘導。
その他の阻害剤:
シクロスポリンA(CsA)やHIF-1α阻害剤は、TGF-β1による線維芽細胞から筋線維芽細胞への変換を抑制。
BETタンパク質阻害剤JQ1は、NOX4/SOD2の不均衡を逆転させ、ROS生成を減少させ、筋線維芽細胞の脱分化を促進。
ボルテゾミブ(BTZ)は、TGF-β1とFGFによって活性化されるキナーゼを抑制し、α-SMAとコラーゲンの発現を大幅に減少。
間葉系幹細胞(MSCs)由来の分泌物成分は、TGF-β Smad2/3経路を選択的に抑制し、筋線維芽細胞の脱分化を促進。
PPARアゴニストは、Smad2/3のリン酸化を抑制し、筋線維芽細胞の脱分化を達成。
Nuclear factor erythroid 2-related factor 2 (Nrf2)
Nrf2は、核因子エリスロイド2類似因子2(NFE2L2)によってコードされ、酸化ストレス応答において重要な転写因子です。Nrf2は、多くの細胞保護遺伝子のプロモーター領域にある抗酸化応答エレメント(ARE)に結合し、酸化ダメージや炎症ストレスから組織や臓器を保護します。Nrf2は抗酸化酵素、免疫および炎症応答の制限因子、組織リモデリングおよび線維化を抑制する遺伝子など、数百の遺伝子の発現を調節します。
NFE2L2はIPFにおける線維芽細胞から筋線維芽細胞への分化(FMD)を抑制し、筋線維芽細胞の脱分化を促進することで線維化の減少に寄与します。siRNAを用いてNrf2をサイレンシングすると酸化ストレスとFMDが誘導されますが、keap1 siRNAでNrf2を活性化すると、α-SMAおよびコラーゲンのRNAレベルでの発現がダウンレギュレートされ、抗酸化防御が強化され、IPF線維芽細胞で筋線維芽細胞の脱分化が促進されます。
亜ヒ酸トリオキシド(ATO)はNrf2活性化剤であり、線維芽細胞における抗酸化遺伝子HO-1の発現を増加させ、TGF-β1によるROS蓄積を減少させます。ATO治療は転写因子NFE2L2の核移行を促進し、Smad2/3のリン酸化を抑制します。スルフォラファン(SFN)もNrf2活性化剤であり、Nrf2の発現を誘導することで肺線維症における筋線維芽細胞の脱分化を促進します。さらに、SFNはIPFにおけるTGF-βの有害な線維促進効果を抑制し、線維芽細胞を制御し、抗酸化防御を回復させます。
したがって、ATOとSFNの両方が線維芽細胞の分化を抑制し、筋線維芽細胞の脱分化を促進することで、肺線維症の治療効果に寄与します。
Ca2+
Ca2+は全身性硬化症(SSc)による肺線維症において重要な役割を果たしており、SScの病理経路の多くがCa2+シグナルと密接に関連しています。SScではTGF-β1の増加が細胞内Ca2+活性と正の相関を持っています。ストア作動性カルシウム流入(SOCE)は炎症および発がんに関連する重要なCa2+シグナルであり、SOCEチャネルは線維芽細胞の増殖、分化、およびECMタンパク質の生成を促進します。SOCE阻害剤は細胞内Ca2+活性を妨げ、SSc筋線維芽細胞を正常な線維芽細胞に脱分化させ、α-SMAおよびフィブロネクチンの発現を減少させます。SOCE関連阻害剤である2-APBは、TGF-β1/SMAD3シグナルを抑制することで線維化マーカー(α-SMA、フィブロネクチン、ビメンチン)を抑制し、筋線維芽細胞の脱分化を誘導します。2-APBの皮下注射はブレオマイシン誘発性の皮膚および肺線維症を改善します。他のSOCE阻害剤であるSKF96365およびインドメタシンも部分的に同様の効果を示します。
KCa3.1はCa2+活性化K+チャネルで、細胞活性化中のカルシウムシグナルと膜電位を調節します。KCa3.1は、線維芽細胞を含む様々な細胞の増殖、活性化、移動、およびメディエーターの放出を調節します。KCa3.1の発現増加は肺線維芽細胞の増殖と機能を促進します。KCa3.1活性はSmadのリン酸化を強化し、TGF-β1に応じてα-SMAのアップレギュレーションを促進します。IPFの大型動物モデルでは、KCa3.1チャネルのブロックが一次羊肺線維芽細胞の線維促進効果を抑制し、初期段階のブレオマイシン誘発性肺線維症を軽減します。ヒト肺線維芽細胞において、KCa3.1活性はIPF由来の肺線維芽細胞の機能を促進します。KCa3.1をブロックすることで、TGF-β1依存性のコラーゲン分泌が減少します。細胞外Ca2+を妨げるか、選択的KCa3.1阻害剤(TRAM-34、ICA-17,043)を使用してKCa3.1イオンチャネルをブロックすると、Smad2/3の核移行が著しく減少し、SMAタンパク質の発現が減少し、ストレスファイバーの形成が阻害されます。これにより、IPF由来のヒト肺筋線維芽細胞(HLMF)が静止した線維芽細胞の表現型に脱分化します。したがって、選択的KCa3.1阻害剤はIPFの新しい治療法を提供する可能性があり、関連する薬剤のさらなる開発が待たれています。
Hypoxia-inducible factor (HIF)-1α
低酸素誘導因子(HIF)-1αは、低酸素条件下での細胞代謝、炎症、および腫瘍形成の重要なメディエーターとして機能します。特発性肺線維症(IPF)患者では、HIF-1αが肺で高発現しており、肺胞上皮細胞(AECs)において小胞体(ER)ストレスを誘発します。また、HIF-1αはAECsにおける上皮間葉転換(EMT)を促進し、TGF-β1による線維芽細胞から筋線維芽細胞への変換(FMD)に関与します。
シクロスポリンA(CsA)は強力な免疫抑制サイクリックノナペプチドであり、間質性肺炎、シェーグレン症候群、IPFの急性増悪などの治療に有効であることが示されています。これらの知見に基づき、CsAの肺線維症に対する抑制効果が探求されています。CsAはHIF-1αタンパク質の分解を促進することで、TGF-β1による線維芽細胞から筋線維芽細胞への変換を抑制することができます。さらに、CsAおよびHIF-1α阻害剤(HIFi)は、肺線維症患者由来の筋線維芽細胞様細胞(MyoLCs)のα-SMAおよびフィブロネクチンの発現レベルを低下させ、脱分化を誘導します。しかし、CsAの強い毒性があるため、その臨床利用にはさらなる探求と評価が必要です。
BET protein
ブロモドメインおよびエクストラターミナルドメイン(BET)タンパク質(Brd2、Brd3、Brd4、BrdT)は、保存されたブロモドメインを通じてヒストンおよび非ヒストンタンパク質上のアセチル化リシン残基に結合し、エピジェネティックリーダーとして機能します。これにより、転写活性化因子や抑制因子をリクルートし、遺伝子発現を調節します【68】。BETタンパク質は、癌や肺線維症の治療標的として有望です。肺線維症モデルでは、TGF-β1による刺激後、Brd4はインターロイキン-6(IL-6)、α-平滑筋アクチン(α-SMA)、およびプラスミノーゲンアクチベーターインヒビター-1(PAI-1)のプロモーターに結合し、線維化を促進します【69】。さらに、肺線維症の病理進行中、TGF-βはNADPHオキシダーゼ-4(NOX4)を増加させ、スーパーオキシドジスムターゼ(SOD2)発現を抑制し、ROS生成を増加させ、筋線維芽細胞の分化を促進します。BETタンパク質阻害剤は、TGF-βによるNOX4/SOD2の不均衡とNrf2の不活性化を逆転させ、ROS生成を抑制し、筋線維芽細胞の分化を逆転させることで、線維症の潜在的な治療法を提供します【43, 44】。
JQ1は小分子阻害剤で、BETタンパク質のブロモドメインとアセチル化リシンの結合を妨げます【68】。JQ1はNOX4/SOD2の発現とNrf2活性に影響を与えて酸化還元バランスを回復し、筋線維芽細胞の再プログラム化や脱分化を促進し、抗線維化効果を発揮します【43】。鈴木健一らの研究はこれを支持しており、JQ1が重度の線維症肺からの初代筋線維芽細胞でα-SMAとED-Aフィブロネクチンの発現レベルを顕著に低下させ、IPF筋線維芽細胞の脱分化に関連するトランスクリプトームスペクトルの包括的な分析を初めて行いました。しかし、BET阻害による筋線維芽細胞の脱分化と関連する遺伝子発現の変化の正確なメカニズムは未解明であり、IPF筋線維芽細胞の脱分化に関連する転写プロファイルは不明のままです【70】。
MSC extracellular vesicles
間葉系幹/間質細胞(MSCs)は、自己再生能力と多分化能を持つ成人幹細胞で、強力な免疫調節、抗炎症、および抗線維化特性を示します。研究によれば、ヒト骨髄由来のMSCsから得られる細胞外小胞(EVs)やエクソソームが、ブレオマイシン誘発性肺線維症を予防および改善することが示されています。これらのEVsは、単球の表現型を全身的に調節し、肺の形態改善、コラーゲン沈着の減少、および肺構造の回復を達成します【71】。別の研究では、非接触型トランスウェル共培養システムで、MSCsとTGF-β誘導性筋線維芽細胞を共培養することで、筋線維芽細胞におけるTGF-β-SMAD2/3シグナル経路の選択的抑制が明らかになりました。この抑制により、筋線維芽細胞は可逆的に線維芽細胞様細胞集団に脱分化しました。これらの線維芽細胞様細胞はTGF-βに感受性を持ち、再び筋線維芽細胞に誘導されることが可能です【46】。MSCが分泌する成分、特にMSC由来の細胞外小胞(MSC-EVs)は、アポトーシスに影響を与えることなく、筋線維芽細胞および線維芽細胞活性化タンパク質アルファ前駆細胞(FAPa + 前駆細胞)の数を減少させました。この減少は、おそらくmiR-29cおよびmiR-129の細胞間移行によって媒介されている可能性があります【14】。
PPAR
PPARは、脂肪組織における脂肪酸貯蔵とグルコース代謝に重要な役割を果たすII型核受容体です。PPARはインスリン感受性を高め、アディポネクチンの放出を促進することで、脂質の取り込みと合成代謝を強化します。このインスリン感受性効果は血糖コントロール療法で広く利用されています【72】。さらに、PPARは抗線維化特性も持ちます。PPAR-γが欠如すると、ヒトおよびマウスの肺線維芽細胞でコラーゲン沈着、SMAD2/3のリン酸化、およびα-SMAレベルが増加します【48】。PPAR-γアゴニスト、特にピオグリタゾンに関する研究は、肺動脈高血圧症(PAH)や慢性線維増殖性腎疾患を抑制または逆転させる経路を明らかにしています【72】。長期のピオグリタゾン治療は、TGFβトランスジェニックマウスの腎臓で転写因子STAT3とEGR1の活性化を抑制し、TGFβ誘発性腎線維症を防ぎます【49】。広く使用されている抗糖尿病薬メトホルミンは、α-SMAとコラーゲンの発現を減少させ、PPARシグナルの活性化を通じてブレオマイシン誘発性肺線維症を緩和します【73,74,75】。
硝酸化脂肪酸(NFAs)は、PPARの独特の内因性活性化剤として機能します【76】。NFAsは、PPARのアップレギュレーション、TGFシグナルおよび作用のブロック、TGFの不活性モノマーへの変換、コラーゲン標的因子MFG-E8のアップレギュレーション、および肺胞マクロファージを刺激してコラーゲンを取り込み分解させることで、筋線維芽細胞の脱分化を促進する可能性があります。実験的証拠は、NFAsが既存の筋線維芽細胞の分化とコラーゲン沈着をマウスモデルの肺線維症で逆転させることを示しています【48】。
ヨモギ由来の脂溶性フラボノイドであるユパチリンは、抗アポトーシス、抗酸化、および抗炎症効果を持つPPAR-αアゴニストとして作用します【77, 78】。ユパチリンはTGF-βで刺激された病原性筋線維芽細胞を直接標的とし、ミオシンの急速な解体を誘導します。この作用により、潜在的なTGF-β複合体が分解され、複数のEMT遺伝子の誘導が抑制されます。さらに、ユパチリンはSmad3のリン酸化を阻害することで、筋線維芽細胞の中間細胞型への脱分化を促進し、線維症を逆転させる可能性があります【47】。
MEK/ERK pathway
マイトジェン活性化プロテインキナーゼ(MAPK)/MAPKキナーゼ(MEK)/細胞外シグナル調節キナーゼ(ERK)シグナルカスケードは腫瘍形成と密接に関連しており、線維症に関連する細胞プロセスを促進する重要な役割を果たします。これらのプロセスには、細胞成長、増殖、移動、アポトーシスの防止、および筋線維芽細胞への変換が含まれます。MEKの阻害は、TGF-αによって誘導される肺細胞の増殖を抑制し、in vivoでのマトリックス遺伝子の発現を減少させることで、TGF-α誘発性の肺線維症を防ぎ、既存の肺線維症の進行を止める効果が示されています。成長因子やマイトジェンはこの経路を通じて信号を伝達し、細胞機能に重要なプロセスでの遺伝子発現を調節します。このシグナルカスケードは腫瘍形成だけでなく、線維症に関連するさまざまな細胞イベントにも関与しており、線維性疾患の予防および治療のための潜在的な治療標的となります。
FGF
線維芽細胞成長因子(FGF)は、細胞核に信号を伝達する重要な成長因子であり、FGF受容体(FGFR)に結合して様々なシグナル伝達経路を活性化します。これらの経路には、Ras、MAPKs、ERKs、Src、p38 MAPKs、ホスホリパーゼCγ(PLCγ)、Crk、jun N末端キナーゼ(JNK)、およびプロテインキナーゼC(PKC)が含まれます。これらの経路は、ETV4およびETV5などの転写因子を活性化します。
TGF-β1はII型肺胞上皮細胞におけるFGF-2の発現と放出を誘導し、FGF-2は抗体中和を通じてTGF-β1によって刺激された線維芽細胞の増殖を抑制します。FGFR1シグナル伝達は、特発性肺線維症(IPF)における線維芽細胞の移動にも重要です。IPF患者の気管支肺胞洗浄液(BAL)中、IPFの肺肥満細胞、ブレオマイシン処理されたマウスの肺マクロファージおよび肥満細胞でFGF2の高レベルが検出されています。
興味深いことに、内因性のFGF2は線維症の生成に必要ですが、TGF-β1による線維性分化には関与しません。逆に、誘導されたFGFの過剰発現および外因性FGFは、I型コラーゲン、α-SMA、ストレスファイバー、およびHsp47シャペロンタンパク質の発現を抑制し、マトリックスメタロプロテイナーゼの発現を増加させることで、TGF-βによる一部の線維促進効果に対抗します。
FGFは様々な臓器および細胞タイプにおいて、ERK、焦点接着キナーゼ、Nkx2.5/Csx、let-7 miRNA、TGFβ1およびTGFβR1のダウンレギュレーション、Smad2リン酸化の減少など、複数のメカニズムを通じてTGF-β1を抑制する役割を果たします。FGFとTGF-β1の複雑な相互作用は、線維症プロセスの複雑さを強調し、治療介入の潜在的な道を開きます。
cAMP/PKA pathway
サイクリックAMP(cAMP)/プロテインキナーゼA(PKA)カスケードは、重要なシグナル伝達経路です。プロスタグランジンE2(PGE2)、β2-アドレナリン受容体作動薬、およびグルカゴン様ペプチド-1(GLP-1)などのさまざまなホルモンは、Gタンパク質共役受容体(GPCR)に結合することによってアデニリルシクラーゼ(AC)を活性化し、ATPをcAMPに変換する反応を触媒します。cAMPはその後、PKAまたはcAMPによって活性化される交換タンパク質(Epac)を活性化します。PKAの活性化は、その主要基質であるcAMP応答エレメント結合タンパク質(CREB)のリン酸化を引き起こします。プロスタグランジン、特にプロスタグランジンE2(PGE2)は、この経路の活性化を通じて、線維芽細胞の分化および筋線維芽細胞の脱分化の調節に重要な役割を果たします。cAMP/PKAカスケードによって組織される複雑なシグナル伝達イベントは、細胞プロセスにおけるその重要性を強調しており、線維芽細胞の動態に関与する調節メカニズムの分子基盤を理解するための基礎を提供します。
PGE
プロスタグランジンE2 (PGE2) は、アラキドン酸から生成される生体内のバイオアクティブな脂質メディエーターで、主にEP2受容体を活性化します。この活性化により、線維芽細胞の増殖、α-SMAおよびコラーゲンの発現、および筋線維芽細胞の分化が抑制されます。これらは細胞内のcAMPの増加を通じて、PKAまたはEpacを活性化することで行われます。ヒト肺線維芽細胞において、Epacの活性化は抗増殖効果をもたらし、PKAの活性化はコラーゲンの発現と筋線維芽細胞の分化を抑制します。
最近の研究は、PGE2の筋線維芽細胞の脱分化における重要な役割を強調しています。PGE2はEP2/cAMP/PKA経路を通じて筋線維芽細胞の脱分化を誘導し、それにより増殖能力を抑制し、アポトーシス感受性を回復させます。PGE2はTGF-β1によって変化した多くの遺伝子発現を逆転させ、これにより細胞接着、収縮性繊維、および細胞移動に影響を与えます。この脱分化は可逆的であり、TGF-βの誘導により再び筋線維芽細胞に分化します。これはFAKシグナル伝達の抑制と関連しています。
さらに、合成プロスタグランジンE1(アルプロスタジル)は、EP2およびEP4受容体を活性化することで肺線維症を軽減または逆転させる可能性が示されています。
他の治療ターゲットと薬剤
【BTZ】
- ボルテゾミブ (BTZ) は、20Sコアプロテアソームのキモトリプシン様活性の可逆的阻害剤です。
- TGF-β1を阻害することで、肺、肝臓、腎臓の線維化を防ぐ能力があります。
- 最近の研究では、BTZがTGF-βとFGF-2によって活性化される重要なキナーゼを阻害し、線維芽細胞の増殖と分化を抑制することが明らかになっています。
- BTZは、MyoFibsおよびIPF Fibsの脱分化を促進し、それらをFAS媒介アポトーシスに対してより感受性を持たせます。
- TGF-β前処理後48時間で、MyoFibsおよびIPF Fibsのα-SMAおよびCol1a2の発現が大幅に減少します。
- 抗線維化効果は、プロテアソーム阻害とは無関係で、二重特異性プロテインホスファターゼ1 (DUSP1) の誘導と活性化に関連しています。
- BTZの肺線維症に対する研究は限られており、その安全性と有効性を評価するためにはさらなる調査が必要です。
【MyoD】
- MyoDは、筋肉の成長と分化に重要な筋原性調節因子です。
- 組織修復と線維化において、筋線維芽細胞の存在と関連しています。
- MyoDはTGF-β1誘導筋線維芽細胞の分化を媒介し、その内因性のダウンレギュレーションは筋線維芽細胞の脱分化と増殖を調節します。
- ERK1/2 MAPKシグナル経路がミトゲンによるMyoDのダウンレギュレーションを媒介します。
- MyoDのサイレンシングは、肺線維症の治療法となり得ます。
- 非老化肺筋線維芽細胞は脱分化する能力がありますが、老化およびIPF筋線維芽細胞はMyoDの持続的なアップレギュレーションによりそれができません。
- MyoDの遺伝子サイレンシングは、IPF筋線維芽細胞のアポトーシス感受性と脱分化能力を回復させます。
【SET8】
- SET8 (PR-set7, SETD8, KMT5A) は、ヒストンH4のリジン20のモノメチル化を特異的に触媒する唯一のリジンメチルトランスフェラーゼです。
- ヒストン修飾タンパク質および非ヒストン因子(p53、Twist、Wnt)と相互作用します。
- 細胞周期、DNA修復、遺伝子転写、およびアポトーシスなどの重要な生理過程に関与しています。
- BLM誘導肺損傷において、SET8はα-SMA陽性細胞の核に局在します。
- SET8の阻害は、筋線維芽細胞におけるα-SMAおよびED-Aフィブロネクチンの発現を著しく抑制します。
- UNC0379はエピジェネティックモジュレーターであり、SET8を阻害し、筋線維芽細胞のα-SMAおよびED-Aフィブロネクチンの発現を減少させ、脱分化を誘導します。
- UNC0379は炎症反応に影響を与えずに肺線維症を部分的に緩和します。
【力の調整】
- 従来の見解では、組織再構築と創傷治癒後、筋線維芽細胞は通常アポトーシスと除去を受けます。
- 最近の研究は、筋線維芽細胞が機械的環境からの情報に基づいてその機能を終える可能性を認識し始めました。
- ECMは筋線維芽細胞の分化の原因であり結果でもあります。
- ECMの硬さは線維芽細胞の持続的な活性化と分化を促進します。
- 機械的ストレスやマトリックスの硬さは、線維芽細胞から筋線維芽細胞への分化を誘導します。
- 機械的ストレスや硬さを減少させると、アポトーシスを誘導し、筋線維芽細胞の収縮性とα-SMAの発現を減少させることができます。
- 境界の硬さを変えることで、筋線維芽細胞の力生成能力を変更し、脱分化またはアポトーシスを引き起こすことができます。
- オメンチン-1は、機械的に活性化された筋線維芽細胞を脂肪線維芽細胞に脱分化させることで線維症の解消を促進し、ECMの除去を助けます。
- 圧力療法は、線維芽細胞から筋線維芽細胞への分化を逆転させ、過剰なコラーゲンの沈着と瘢痕形成を抑制します。
- 機械的ストレスとマトリックス硬さが筋線維芽細胞の脱分化に及ぼす影響について、さらに調査する必要があります。