医薬品等の市販後安全対策としてのGVPの具体的運用方法

1 はじめに
医薬品等の安全性監視活動は,ファーマコビジランス(Pharmacovigilance)と呼ばれ,欧州医薬品審査庁(以下「EMA」という。EMA ; European Medicines Agency)では,ファーマコビジランスについて,「医薬品の使用が承認される前は,その安全性と有効性の根拠は,患者が慎重に選択され,管理された条件下で非常に綿密に追跡される臨床試験の結果に限定されている。これは,医薬品の承認時に,限られた期間,比較的少数の選択された患者で試験されたことを意味する。承認後の薬は多くの患者に長期間,他の薬と一緒に使用される可能性がある。このような状況では,何らかの副作用が発生する可能性がある。したがって,全ての医薬品の安全性は,医療現場での使用を通じて監視されることが不可欠である。」1)と述べている。この考え方は,日米欧を中心に世界各国で適応され,医薬品に留まらず,医療機器,医薬部外品,化粧品,再生医療等製品に当てはまり,ファーマコビジランスを適切に運用する基準が,GVP(GVP ; Good Pharmacovigilance Practice)と称される。
我が国の医薬品等の市販後の安全性監視活動は,平成16年厚生労働省令第135号「医薬品,医薬部外品,化粧品,医療機器及び再生医療等製品の製造販売後安全管理の基準に関する省令」(以下「GVP省令」という。)の基準がある。一方,医薬品等の開発段階では平成9年厚生省令第28号「医薬品の臨床試験の実施の基準に関する省令」(以下「GCP省令」という。GCP ; Good Clinical Practice)の基準があり,厳格に管理された患者等において安全管理情報の収集・評価・対策がなされるため,本誌では市販後について説明する。
なお,本誌では,厚生労働省と独立行政法人 医薬品医療機器総合機構(以下「PMDA」という。PMDA ; Pharmaceuticals and Medical Devices Agency)で使い分けしているが,PMDAには,その設置法で,昭和35年法律第145号「医薬品,医療機器等の品質,有効性及び安全性の確保等に関する法律」(以下「薬機法」という。)の「調査」権限のみが与えられ,個別症例報告はPMDAに対して行うが,薬機法の行政指導は,厚生労働省や各都道府県が担い,例えば通知等は厚生労働省から発出される。
2 我が国のGVP省令の特徴
欧米でのGVP基準と日本のGVP省令は,医薬品規制調和国際会議(以下「ICH」という。ICH ; International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use)での国際調和により概ねハーモナイズされていると考えて良い。なお、本誌では第一種医薬品製造販売業者を製薬企業という。
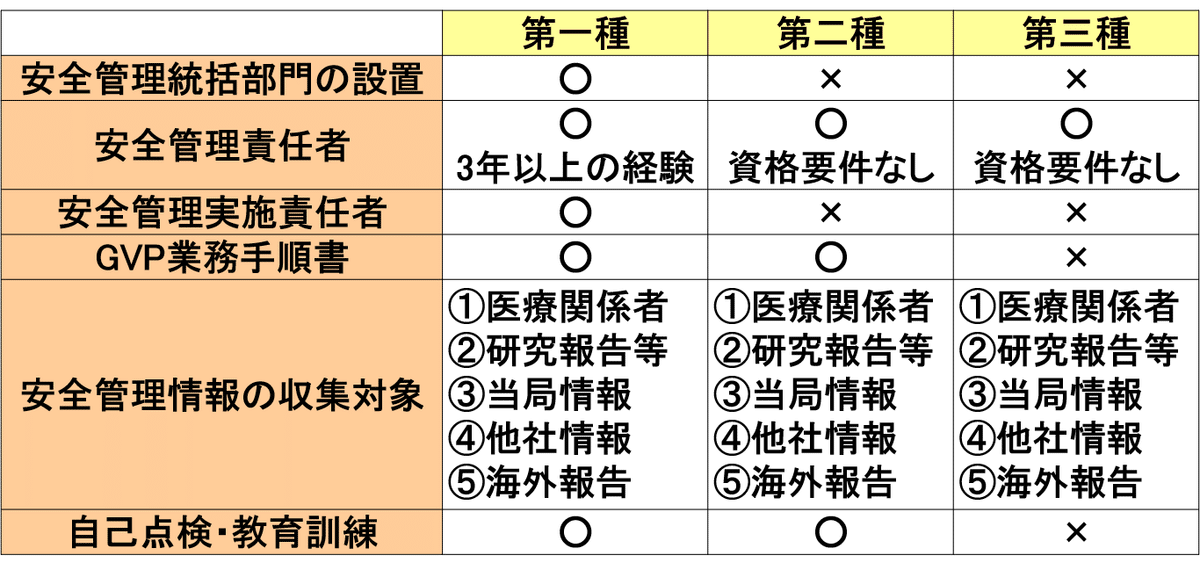
我が国でのGVPの特徴の一つとして製造する製品の種類毎に許可区分が異なるので,表1(医療機器を除く)と表2(医療機器)に示す。
また,製造販売業許可毎のGVP省令の要件を表3に示す。


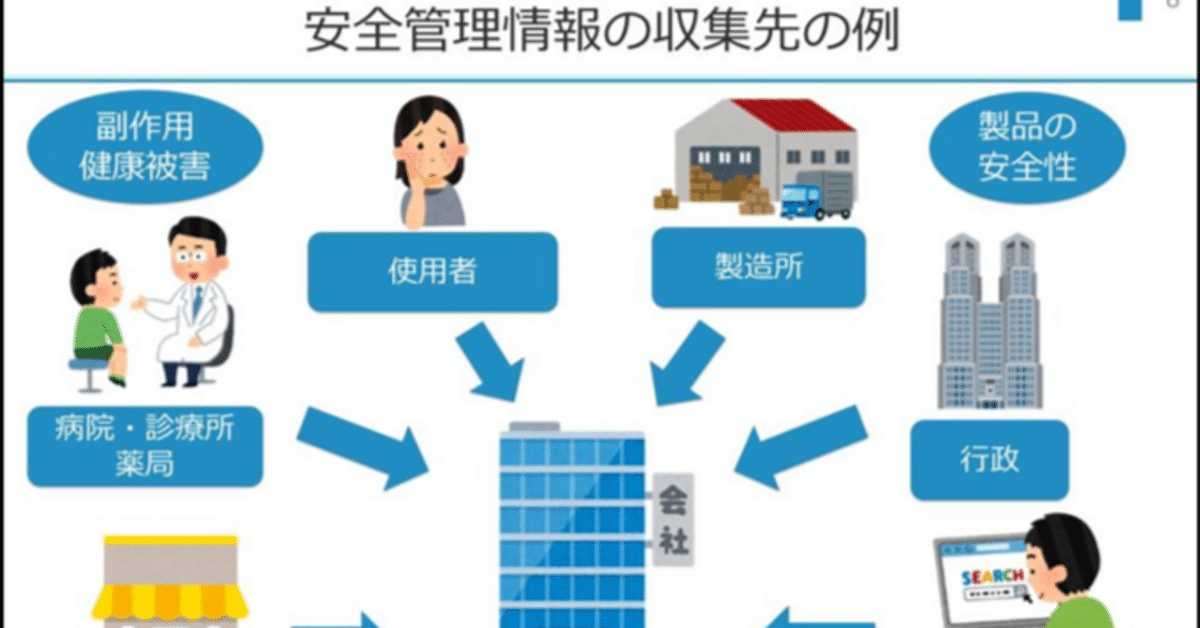
3 安全管理情報の収集
製薬企業では,その会社製品の安全管理情報の収集義務があり,必ずしも安全性に限っていない。例えば,文献等も含め安全性に限らず,有効性や品質も含めて,常に情報を入手する必要がある。なお,「安全管理情報」とはGVP省令第2条で「医薬品,医薬部外品,化粧品,医療機器又は再生医療等製品の品質,有効性及び安全性に関する事項その他医薬品等の適正な使用のために必要な情報をいう。」と規定されている。
これらの情報源としては,以下のとおり多義に亘る。
1)国内における医療関係者からの報告
2) 国内外で収集される学会報告・文献報告・その他研究報告に関する情報
3) 厚生労働省その他政府機関,都道府県及び医薬品医療機器総合機構からの情報
4)外国政府,外国法人等からの情報
5)他の製薬企業等からの情報
4 有害事象と評価
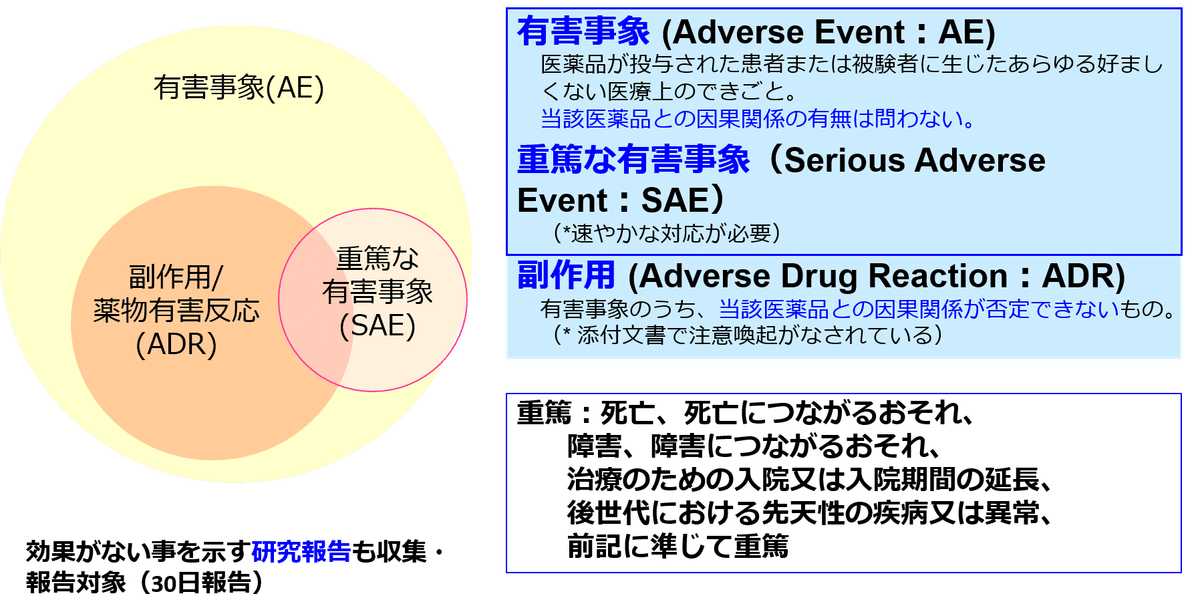
「有害事象」とは,医薬品等の服用後に起きた,あらゆる健康上の問題であるので,市販後に収集されるものは有害事象となる。 例えば, 薬を服用し,交通事故に遭ってしまったら有害事象として取り扱う必要がある。このように有害事象は,医薬品等との因果関係が明らかなものだけでなく,因果関係が,否定できないもので,未知のもの,不明なものも含む。一方,副作用とは,医薬品等による有害事象のうち,本来の作用以外の作用のことで,具体的には添付文書に記載のある「副作用」や使用上の注意に記載されているもの等で,図1に示す。
欧米における製薬企業の有害事象の収集対象は,特別な状況(Special Situation)についても対象となるので,図2に示す。

5 情報入手日
有害事象の入手に際し,最低限の4情報(①患者識別情報(イニシャル,年齢,性別等),②被疑薬名(自社被疑薬),③情報源(医師,薬剤師,消費者等),④有害事象名・感染症名(海外では「特別な状況」も))が収集された際にPMDA報告義務が発生し,情報入手日としてのタイム・クロック(入手日を0日目とする)が動き出す。
例えば,コールセンター業務を委託している場合,委託先の受付担当者が上記情報を入手した時点でタイム・クロックが動き出す。なお,この例示の様に,製薬企業が一部のPV業務を委託する場合は,企業間における安全管理情報の取扱いについての契約(所謂PVA ; Pharmacovigilance Agreement)を締結する必要がある。
更に,製薬企業では,より詳細で正確な安全管理情報を得るために,4情報の収集に限らず,有害事象が生じた際の状況や併用薬剤等をできる限り収集する追加調査(follow-up)を行い,適正な症例評価に資する安全管理情報の入手に勤める必要がある。
6 有害事象の収集・報告・解析におけるMedDRA
製薬企業などでは有害事象を医師等から報告を受けるが,医師により言い回しが異なる場合や,日本語(漢字や仮名混じり等)で,そのままでは細か過ぎるなど有害事象の集積件数や統計解析には不向きである。 そのため,報告された有害事象をMedDRA(MedDRA ; Medical Dictionary for Regulatory Activities)によって,コーディング(数字に置き換え)する必要がある。
MedDRAとは,医薬品等の登録,記録文書および安全性監視における規制上のコミュニケーションに用いる国際的医学用語集(有害事象分類辞書)であり,医薬品の規制当局と製薬企業において医薬品の市販前および市販後の両規制段階において臨床データの入力,検索,評価および提示に使用される。MedDRAのコーディングによって,多言語間でも有害事象の集積・解析等が可能となる。なお,MedDRAは,概ね半年に1回の頻度で更新される。
我が国の製薬企業では, 医薬品副作用 ・感染症報告(法第68条の10),感染症定期報告(法第68条の14),治験薬副作用・感染症症例報告(法第80条の2第6項),安全性定期報告(薬機法施行規則第63条)などの報告において,有害事象の集積数の報告が求められている。
MedDRAの階層構造を図3に示す。医師等からの報告は,下層語(LLT)の中から最も近いものを選ぶ。それに対応する上位の基本語(PT)で集計する。

7 重要な医療事象(IME)リスト
重要な医療事象(IME ; important medical event)リストは,EudraVigilance専門家作業部会(EV-EWG)が定めたもので,「欧州連合(EU)における関係者の日常的なPV活動の枠内で,疑われる副作用の分類,集計されたデータ分析及び症例評価を容易にすることを目的としている。」2)とされている。運用面では,症例の評価者によって曖昧な重篤性の判断基準を,IMEとして定義したものであり,IME リストに入る有害事象は,報告医が非重篤として認識していても,欧州の規制当局に報告する際は,重篤なものとして取り扱う。白血球数減少を例に取れば,白血球減少症(Leukopenia)とコーディングされればIMEで「重篤」とされるが, 白血球数減少 (White blood cell count decreased)とコーディングされれば,報告医が非重篤とした場合,非重篤が許容される。なお,報告医の重篤度を製薬企業がダウングレードする事は許容されない。
8 症例の評価
製薬企業での症例の評価は,重篤性,予測性,因果関係の3つのパラメータで評価を行う。
重篤性について,薬機法施行規則第228条の20又は第273条において,死亡,障害,死亡又は障害につながるおそれのある症例,治療のために病院又は診療所への入院又は入院期間の延長が必要とされる症例,これらに準じて重篤である症例,後世代における先天性の疾病又は異常は,重篤とされている。
予測性について,「使用上の注意等から予測することができないもの」は予測性が無い(以下「未知」という。)とされる。これは注意事項等情報における「使用上の注意」(「警告」, 「重要な基本的注意」, 「相互作用」,「副作用」等)に記載されていないもの,あるいは,記載されていてもその性質又は症状の程度,特異性等が記載内容と一致しないものは未知であると厚生労働省から通知3)されている。
因果関係について,「副作用によるものと疑われるもの」に因果関係があるとされるが,因果関係が否定できるもの以外のものを指し,因果関係が不明なものも含まれることとも報告対象になると通知3)されている。
因果関係は,JCOGにおける因果関係の程度の分類(①definite(certain):明確に,②probable:おそらく,③possible:ありうる,④unlikely:ありそうにない,⑤not related(unrelated):関係ない,⑥unassessable(conditional):評価不能)4)が示されているので,参考にされたい。
なお, 重篤性,予測性,因果関係の関係から報告期限が,薬機法施行規則第228条の20又は同施行規則第273条に規定されており,報告遅延や報告漏れは処罰の対象となる。
9 市販直後調査と市販後調査について
新医薬品が一旦,販売開始されると,治験時に比べてその使用患者数が急激に増加するとともに,使用患者の状況も治験時に比べて多様化することから,治験段階では判明していなかった重篤な副作用等が発現することがある。
「市販直後調査」とは,欧米に無い我が国の市販後安全対策であるが,製薬企業が販売を開始した時点から6か月間,医療機関に対し確実な情報提供,注意喚起等を行い,医薬品の適正な使用に関する理解を促すとともに,当該医薬品の副作用によるものと疑われる症例等の発生を迅速に収集し,必要な安全対策を実施して副作用等の被害を最小限にすることを主な目的とする調査で,図4に示す。なお,本調査は医療機関に対し適正使用の情報提供,注意喚起等を行い,医薬品の適正な使用を促すことから,GVP省令の規制下で行われる。
対象となるのは,新医薬品である。ただし,実施しない合理的な理由がある場合は対象とならない場合もある。
これに対し,製造販売後調査とは,一般に「市販直後調査」の後,行われる調査で,我が国では以下の表4に分類され,平成16年厚生労働省令第171号「医薬品の製造販売後の調査及び試験の実施の基準に関する省令」通称GPSP省令(GPSP;Good Post-marketing Study Practice)の下で実施される。
一方,欧州における製造販売後調査とは,Post-authorisation safety studies (PASS)といい(臨床試験または非介入研究のいずれかも含む) という概念で,我が国のGPSP省令より幅広く,例えば,便秘に関する調査を,大衆に行う場合であって,必ずしも自社薬に限らず,服用している便秘薬を聞く欄と,また,お通じに関する自由記載欄があった場合には,自由記載欄から有害事象を収集する必要がある。


10 安全確保措置について
このように個別症例の収集や調査・研究で収集された有害事象は,その医薬品等の安全確保措置に活かされなければいけない。製薬企業では,欧米では主に統計解析によるシグナルとして認められた有害事象を「企業中核データシート」(CCDS ; Company Core Data Sheet)に掲載し,医療関係者等に注意喚起して安全に使用していただくこととなる。一方で,我が国は収集された1症例ごとに,重篤性,予測性,因果関係を丁寧に評価して,安全対策に役立てることが多い。また,安全対策は製薬企業が規制当局と調整の上,適切な内容で講じられる。具体的には,添付文書改定,緊急安全性情報,安全性速報等と言ったもので広く医療関係者等に周知して,医薬品等の適正使用に供される。
1)GVP省令での安全確保措置
廃棄,回収,販売の停止,添付文書の改訂,MR等による医療関係者への情報の提供,その他と定められている。(GVP省令第8条)
2)緊急安全性情報,安全性速報5)について
緊急安全性情報は,医薬品等について,以下に掲げるいずれかの状況からみて,国民(患者),医薬関係者に対して緊急かつ重大な注意喚起や使用制限に係る対策が必要な状況にある場合とされている。
・ 副作用・不具合等の報告における死亡,障害若しくはこれらにつながるおそれのある症例又は治療の困難な症例の発生状況
・ 未知重篤な副作用・不具合等の発現など安全性上の問題が有効性に比して顕著である等の新たな知見
・ 外国における緊急かつ重大な安全性に関する行政措置の実施
・ 緊急安全性情報又は安全性速報等による対策によってもなお効果が十分でないと評価された安全性上の問題
安全性速報は,保健衛生上の危害発生・拡大の防止のため,緊急安全性情報に準じ,医薬関係者に対して一般的な使用上の注意の改訂情報よりも迅速な注意喚起や適正使用のための対応(注意の周知及び徹底,臨床検査の実施等の対応)の注意喚起が必要な状況にある場合とされている。
3)添付文書の改訂について
安全確保措置を適切に周知するためには,厚生労働省から通知により添付文書の改訂内容が指示され,広く医療機関,薬局等に速やかに伝達されることによってなされる。
11 医薬品リスク計画(RMP)の概要
医薬品の安全性の確保を図るためには,開発の段階から市販後に至るまで常にリスクを適正に管理する方策を検討することが重要である。医薬品リスク管理計画(以下「RMP」という。RMP ; Risk Management Plan)は,医薬品の開発から市販後まで一貫したリスク管理をひとつの文書に分かり易くまとめ,調査・試験やリスクを低減するための取り組みの進捗に合わせて,または,定期的に確実に評価が行われるようにするものである。
また,RMPを公表して,医療関係者と市販後のリスク管理の内容を広く共有することで,市販後の安全対策の一層の充実強化が図られる。
製薬企業がRMPを作成する上で,日本人に特有な未知・重篤症例で発生頻度,発生傾向から安全性検討事項に加えるべき事例を紹介する。例えば,抗がん剤による薬剤性間質性肺炎・肺障害の発生率は日本人と外国人の比較で,「日本人は薬剤性肺障害を起こしやすい」ことが疫学的調査からと論じられているので表5に示す。
PMDAに報告された有害事象はPMDAのホームページからダウンロードが可能であり,収集された有害事象の内訳割合にはバイアスが入り難い事から,類似の他剤との比較で,自社薬での特定の有害事象の発生傾向の検出に役立つことから,PMDAでは製薬企業に積極的な利活用を求めている。


12 定期的ベネフィット・リスク評価報告(PBRER)について
市販後の医薬品のベネフィットとリスクに関する情報をICH E2C(R2)6)での合意に基づき,製薬企業に対して「定期的ベネフィット・リスク評価報告(PBRER ; Periodic Benefit-Risk Evaluation Report)」として取りまとめ,年に1回,厚生労働省に報告する等により,市販後の安全対策が通知7)で求められている。
ICH E2C(R2)でのPBRERの主たる目的は,「製品の全体的なベネフィット・リスクプロファイル評価を可能にするため,医薬品のリスク及び承認適応に対するベネフィットに関する新しい情報又は明らかになりつつある情報の包括的,簡潔かつ重要な分析を示すことにある」とされている。PBRERには,以下の累積情報に基づき,調査期間に製薬企業が入手した医薬品に関する新規の情報の評価が含められていなければならない。
医薬品のベネフィット・リスクプロファイルに影響を及ぼす可能性がある関連する新たな安全管理情報の要約
調査期間において入手した重要な新しい有効性/有用性の情報の要約
調査期間において製薬企業が入手した情報が,医薬品のベネフィット・リスクプロファイルに関する過去の知見に一致しているか否かの検討
重要な新しい安全管理情報が明らかになりつつある場合には,承認適応に対する総合的なベネフィット・リスク評価の実施
14 PV業務の委受託とPVAについて
我が国では,昭和36年厚生省令第1号「医薬品,医療機器等の品質,有効性及び安全性の確保等に関する法律施行規則」第97条にPV業務を委託する場合,同規則第98条にPV業務の再委託する場合の規定があり,これらに従う必要がある。安全性データベースに安全管理情報を自社の社員で入力を行う製薬企業は少なくなっており,委託している企業が殆どである。また,製薬企業もコールセンターを社内に設置せず,委託契約している場合が多い。このようなコールセンターでも,安全管理情報を入手すれば,入手日が動きだすことから。多くの製薬企業は,これらデータベース入力を外注し,また,その外注先がコールセンター業務を再委託する場合が,これらの規定に該当する。ここで,重要なのが業務委託を外注する場合には,ファーマコビジランス契約(PVA)が不可避となることである。PVAの考えは,欧州のEU-GVP Module I 8)QMS(Quality Management System)に記載されている。
15 コンビネーション製品の副作用・不具合報告について
コンビネーション製品とは,2つ又はそれ以上の医薬品及び医療機器で構成される製品を指して言うが,現在は多くの製品がコンビネーション製品となっており,具体的な品目は通知9)されているので参照戴きたい。身近な代表的事例としては,糖尿病患者様で使用されるペン型の自己注射を思い浮かべて戴けるとイメージしやすいかと思う。
薬機法施行規則第228条の20又は第273条の「不具合」とは,「破損,作動不良等広く品質,安全性,性能等に関する被験機器の具合がよくないことをいい,設計,交付,保管,使用のいずれの段階で生じたものであるかを問わないこと。」と,また,当該不具合によって不具合が発生する「おそれ」とは,「現実には死亡,障害等は発生していないが,死亡,障害等が発生するおそれがあると認められるものであること。」と通知10)されており,PMDAへの報告対象となる。GVP省令では,安全管理責任者は品質保証責任者や他の部署との安全管理情報について相互の連携を手順書して定めるよう求めている。
米国に目を向けると,部門間の情報連携に関し,品質情報を扱うデータベースと安全管理情報を扱うデータベースに同じ情報が入っているかを米国食品医薬品局(FDA ; Food and Drug Administration)の査察では確認されることとなるので注意が必要である。
更に,類似医療機器(similar device)の取り扱いでFDAは類似のdeviceの場合は,不具合報告を求めている11)。例えば,日本であれば,類似のdeviceでも国内製造であればPMDA報告の必要はないが,FDAは免れないので,米国で承認を有する場合は,国内の不具合情報をFDAに報告する必要がある。

☆医療機器部分の不具合(針折れ等)が原因の健康被害であることが明らかであり,医薬品に起因する健康被害(副作用等)がない場合は,不具合報告のみ提出.
☆医療機器部分が原因の健康被害(金属アレルギー等)であることが明らかで,医薬品との関連が否定されている(医薬品による,副作用等がない)場合は,不具合報告のみ提出.
16 おわりに
本誌では医薬品等の市販後安全対策の概要を述べさせて戴いたが,実運用での安全対策は,多くの規制・基準があるので注意して戴きたい。また,医薬品は,機械の様に部分的な改良ができないため,その適正使用によって,重大な副作用が生じないよう,患者様におかれては医師,薬剤師の指導の元,適正に服用・使用して戴く事が重要であること,また,副作用が生じた場合,躊躇なく医師や薬剤師に相談されることを申し添えたい。
参考文献
1)European Medicines Agency, Pharmacovigilance : Overview
https://www.ema.europa.eu/en/human-regulatory-overview/pharmacovigilance-overview
2)Inclusion/exclusion criteria for the “Important Medical Events” list, 18 March 2021, EMA/126913/2021
https://www.ema.europa.eu/en/documents/other/inclusion-and-exclusion-criteria-important-medical-events-list-meddra_en.pdf
3)令和2年8月31日,薬生薬審発0831第12号,薬生安発0831第3号,厚生労働省医薬・生活衛生局医薬品審査管理課長,厚生労働省医薬・生活衛生局医薬安全対策課長通知「E2B (R3) 実装ガイドに対応した市販後副作用等報告及び治験副作用等報告について」
4)JCOG臨床安全管理情報取扱いガイドライン(ver.2.0 改訂日:2009/11/10)
https://jcog.jp/A_020_0010_16_2.pdf
5)令和5年8月10日,薬生安発0810第2号,厚生労働省医薬・生活衛生局医薬安全対策課長通知「緊急安全管理情報等の提供に関する指針について」の一部改正について
6)ICH guideline E2C (R2) on periodic benefit-risk evaluation report (PBRER), January 2013, EMA/CHMP/ICH/544553/1998
7)平成25年5月17日,薬食発0517第2号,厚生労働省医薬食品局長通知「薬事法施行規則の一部を改正する省令の施行及び新医療用医薬品に関する安全性定期報告制度について」
8)Module Ⅰ - Pharmacovigilance systems and their quality systems, 22 June 2012, EMA/541760/2011
9)平成26年10月24日,薬食審査発1024第2号,薬食機参発1024第1号,薬食安発1024第9号,薬食監麻発1024第15号,厚生労働省医薬食品局審査管理課長,同省大臣官房参事官,同省医薬食品局安全対策課長,同省医薬食品局監視指導・麻薬対策課長通知「コンビネーション製品の承認申請における取扱いについて」
10)令和2年8月31日,薬生発0831第9号,厚生労働省医薬・生活衛生局長通知「独立行政法人医薬品医療機器総合機構に対する機械器具等に係る治験不具合等報告について」
11)The product, or a similar product marketed by the applicant, “would be likely to cause or contribute to a death or serious injury if the malfunction were to recur” (21 CFR 803.3 (o)(2)(ii) and 803. 50)