
初心者の菌叢解析 Qiime2で解析(13) PICRUSt2の使い方編

寒さの深まる2月の上旬です。
某ウイルスも過去最大の感染者数記録しており、会いたい人にも会えない日々が続いています。
執筆していた論文が一段落したので、こちらの投稿を再開した次第です。
今回はPICRUSt2での解析を進めていきます。
ここまで来た方にとっては難しくないと思います。
1. はじめに
PICRUSt2はメタゲノム解析で得た配列情報を元に、その機能性を予測するツールです。これにより、16S rRNAの配列に基づく細菌叢の働き(functional potential )を予測できます。
16S rRNAだけで無く、たのマーカー遺伝子でも使用可能です。
そのため、菌叢を「菌種」として捉えるのでは無く、「遺伝子群」として捉えることができます。
PICRUSt2はver.1と比較し、リファレンスゲノム等のデータベースが更新されており、より正確性が増しているようです。
PICRUSt2のインストールは過去の投稿をご確認いただければと思います。
時間があいてしまい、申し訳ありませんでした。
一応参考資料載せておきます。
2.PICRUSt2の確認
まずは、ターミナル(Ubuntu)を起動し、目的ディレクトリに移動後、Qiime2を起動します。
cd /Users/ユーザー名/Desktop/Qiime2_test
conda activate qiime2-2021.2次に以下のコマンドを入力し、「PICRUSt2」がインストールされていることを確認します。
qiime --help
コマンドリストに「PICRUSt2」が無ければインストールできていないことになります。
Qiime2のバージョン等(ver. 2021.2を使用)を再確認し、インストールを行ってください。
3.PICRUSt2解析①
PICRUSt2の解析には基本的にこれまでの解析で取得した2つのファイルが必要です。
1.table.qzaファイル
2.rep-seqs.qzaファイル
この2つのファイルはこれまで解析を行っていれば既に取得しているファイルのはずです。必要な方は以下の投稿をご確認ください。
それではコマンドを入力し、解析を行います。
qiime picrust2 full-pipeline \
--i-table table.qza \
--i-seq rep-seqs.qza \
--output-dir q2-picrust2_output \
--verboseここの解析には時間がかかります。
うまくいけば、コマンドの通り「q2-picrust2_output」というディレクトリが生成されます。
ディレクトリの中には、以下の3つのファイルが保存されていると思います。
1.ec_metagenome.qza
2.ko_metagenome.qza
3.pathway_abundance.qza
次に以下のコマンドでそれぞれの「qzaファイル」を可視化していきます。
qiime feature-table summarize \
--i-table q2-picrust2_output/pathway_abundance.qza \
--o-visualization q2-picrust2_output/pathway_abundance.qzv
qiime feature-table summarize \
--i-table q2-picrust2_output/ec_metagenome.qza \
--o-visualization q2-picrust2_output/ec_metagenome.qzv
qiime feature-table summarize \
--i-table q2-picrust2_output/ko_metagenome.qza \
--o-visualization q2-picrust2_output/ko_metagenome.qzv「qzvファイル」が生成されたら、Qiime2 Viewで内容を確認します。
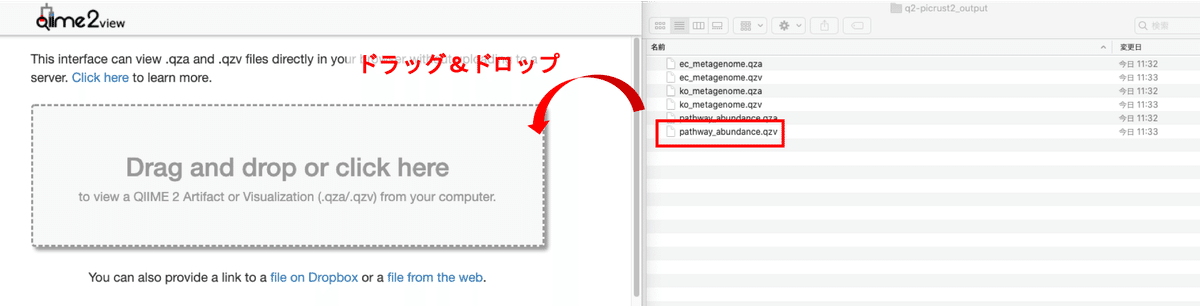
まずは「pathway_abundance.qzv」をQiime2 Viewにドラッグ&ドロップし、Interactive Sample Detailのタブを確認します。
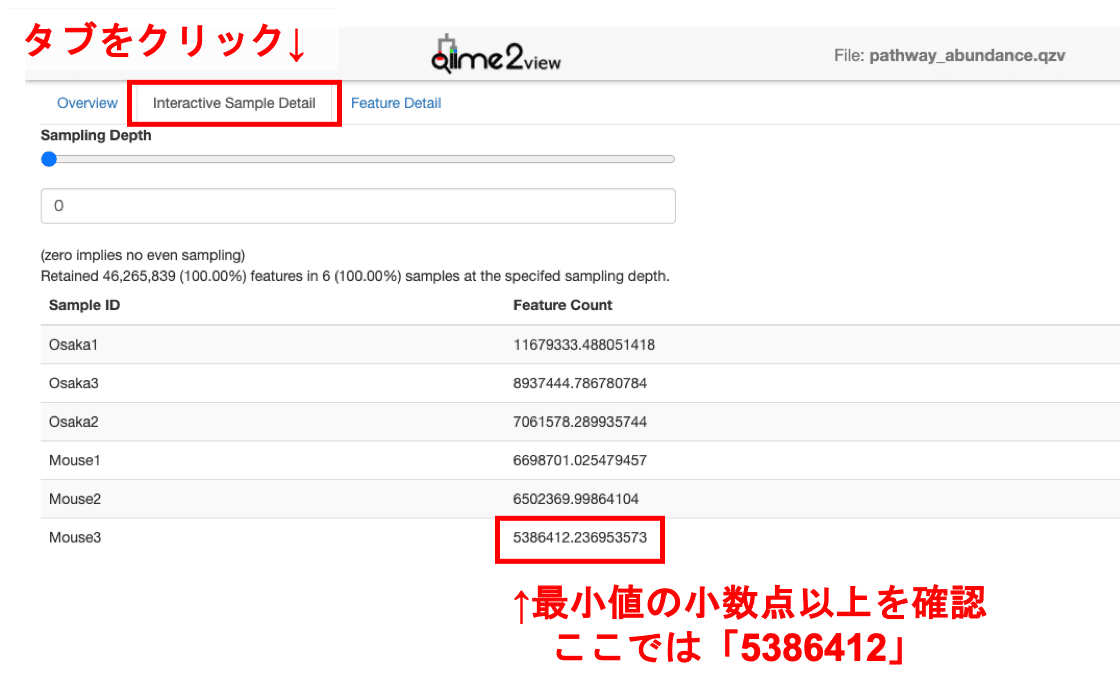
Feature Countの一番下にある最小値(小数点以上)を記録します。
以下のコマンドを実行し、主成分分析を行います。
qiime diversity core-metrics \
--i-table q2-picrust2_output/pathway_abundance.qza \
--p-sampling-depth 5386412 \
--m-metadata-file sample-metadata.txt \
--output-dir q2-picrust2_output/pathabun_core_metrics_out今回はpathway_abundanceの解析をまず行う為、「--i-table」の部分には、「q2-picrust2_output/pathway_abundance.qza」を入力します。
真ん中に「/」があるので、こちらは「q2-picrust2_output」ディレクトリの中の「pathway_abundance.qza」ファイルを指定していることになります。
「--p-sampling-depth」の部分は先ほど確認し、たFeature Countの最小値(小数点以上; 5386412)を入力します。
解析が終了すると、「pathabun_core_metrics_out」というディレクトリが生成され、主成分分析の結果を確認できると思います。
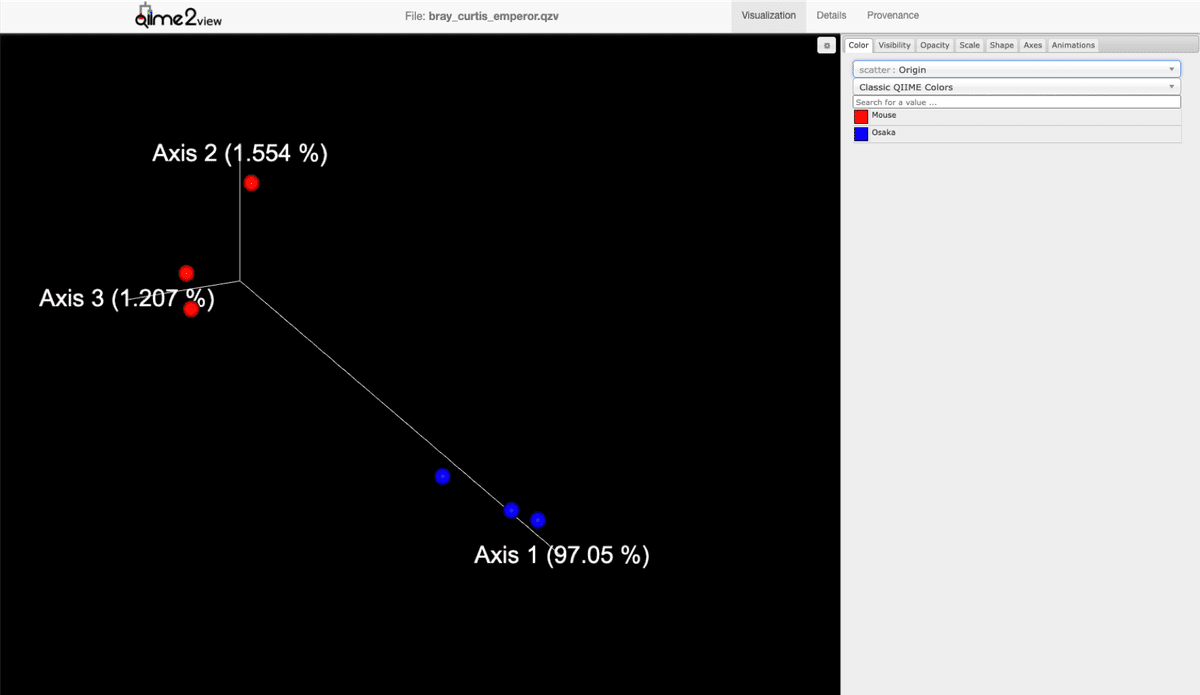
「Mouse; 赤」と「Osaka; 青」は完全に異なるクラスターを形成しています。全く種類の違う環境に住む細菌叢ですので、必要となる遺伝子群も全く異なるのは当たり前ではありますが、このような結果となりました。
ここではPathway_abundanceを中心に解析しましたが、同時に生成される「ec_metagenome.qza」と「ko_metagenome.qza」についても同様に解析可能です。
「--i-table」の部分を目的のファイルに変更し、「--p-sampling-depth」で入れる値を再度確認してください。
4.PICRUSt2解析②
次は具体的な項目を確認する為、表形式でファイルを出力します。
以下のコマンドを実行します。
qiime tools export \
--input-path q2-picrust2_output/pathway_abundance.qza \
--output-path q2-picrust2_output/pathabun_exported
qiime tools export \
--input-path q2-picrust2_output/ec_metagenome.qza \
--output-path q2-picrust2_output/ec_metagenome_exported
qiime tools export \
--input-path q2-picrust2_output/ko_metagenome.qza \
--output-path q2-picrust2_output/ko_metagenome_exported以下のコマンドで、表形式に変換します。
biom convert \
-i q2-picrust2_output/pathabun_exported/feature-table.biom \
-o q2-picrust2_output/pathabun_exported/pathabunーfeature-table.biom.tsv \
--to-tsv
biom convert \
-i q2-picrust2_output/ec_metagenome_exported/feature-table.biom \
-o q2-picrust2_output/ec_metagenome_exported/ec-feature-table.biom.tsv \
--to-tsv
biom convert \
-i q2-picrust2_output/ko_metagenome_exported/feature-table.biom \
-o q2-picrust2_output/ko_metagenome_exported/ko-feature-table.biom.tsv \
--to-tsv生成された「pathabunーfeature-table.biom.tsv」をエクセルで開いて確認してみます。
右に各群の平均値を自分で追加してみました。
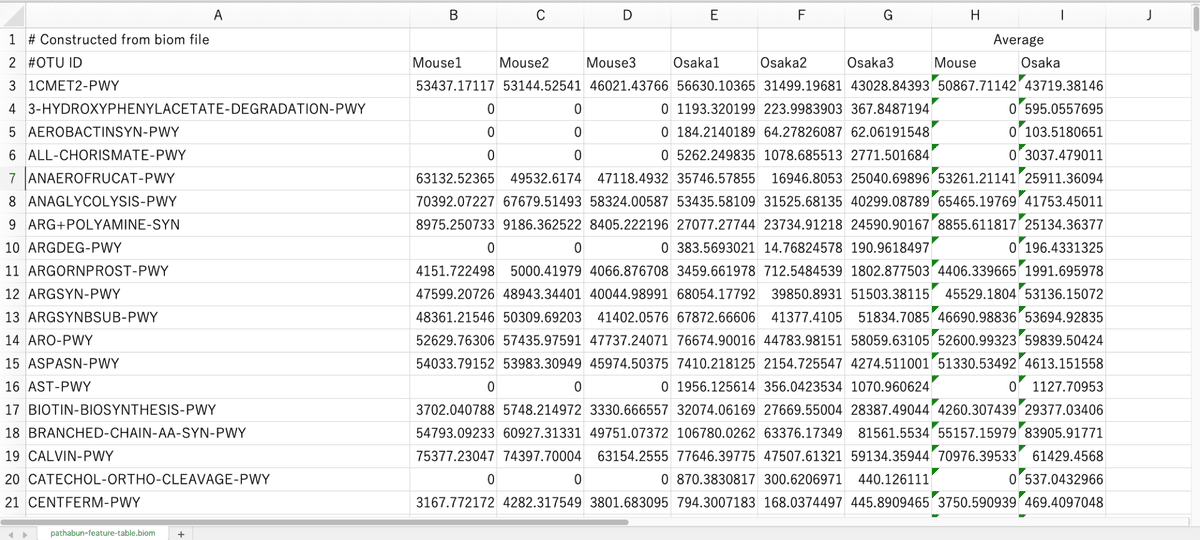
大雑把なP値やlogFCを計算してみたところ、例えばマウスの腸内では「フコースやラムノース」の分解に関するPathwayが検出され、大阪湾の菌叢からは「バイオクロロフィル」の合成に関するPathwayが出て来ました。
それぞれの生育環境を考えれば当然の結果のように思います。
表形式で出力できれば、自分で「FDR」を計算し、logFCを計算すればボルケーノプロット作れると思います。ANCOMでの解析も可能だと思います。
今回は以上になります。
次回はPICRUSt2の解析結果をもとに、KEGG pathwayにマッピングしてみようと思います。