
Photo by
hiroshi_yoshida_
Ensemble ID→Gene IDへの変換方法【RNAseq】

*「ENSMUST」で始まるマウスのEnsemble ID用です。
biomaRtを用いて、Ensenble ID→Gene Symbolへの変更方法をまとめます。
必要なパッケージのインストールとロード
まず、biomaRt パッケージをインストールし、Rセッションにロードします。
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("biomaRt")
library(biomaRt)Ensemblのマートを設定
マウスのデータを取得するために、Ensemblのマートを設定します。
#マウスサンプルの場合
ensembl <- useMart("ensembl", dataset = "mmusculus_gene_ensembl")#ヒトサンプルの場合
ensembl <- useMart("ensembl", dataset = "hsapiens_gene_ensembl")Ensembl IDからSYMBOLへの変換
EnsemblトランスクリプトIDから遺伝子シンボルに変換するために getBM 関数を使用します。
# 'data' データフレームの1列目のIDを取得(例: 'ENSMUST00000115585')
#例「"取り込んだCSVファイルの名前"$"1列目の名前" 」は「human.gene.csv$...1」のように
ensembl_ids <- "取り込んだCSVファイルの名前"$"1列目の名前"
# getBM 関数を使用して変換
symbols <- getBM(attributes = c('ensembl_transcript_id', 'external_gene_name'),
filters = 'ensembl_transcript_id',
values = ensembl_ids,
mart = ensembl)結果を元のデータフレームに統合
getBM で得たデータを元の data データフレームに統合します。
merged_data <- merge(取り込んだCSVファイルの名前, symbols, by.x = "1列目の名前", by.y = "ensembl_transcript_id", all.x = TRUE)このコードでは、data データフレームの "EnsemblID" 列と symbols データフレームの "ensembl_transcript_id" 列に基づいて統合を行います。
結果のCSVファイルとしての出力
最後に、統合されたデータをCSVファイルとして出力します。
write.csv(merged_data, "merged_data.csv", row.names = FALSE)データの整理
次に、RNA chep用に書き出したデータの整理を行う。
まず、書き出した「merged_data.csv」ファイルをエクセルファイルで開き、1番左の列の「Ensemble ID」を消し、代わりに1番右の列の「external_gene_name」が1番左にくるようにカット&ペーストする。
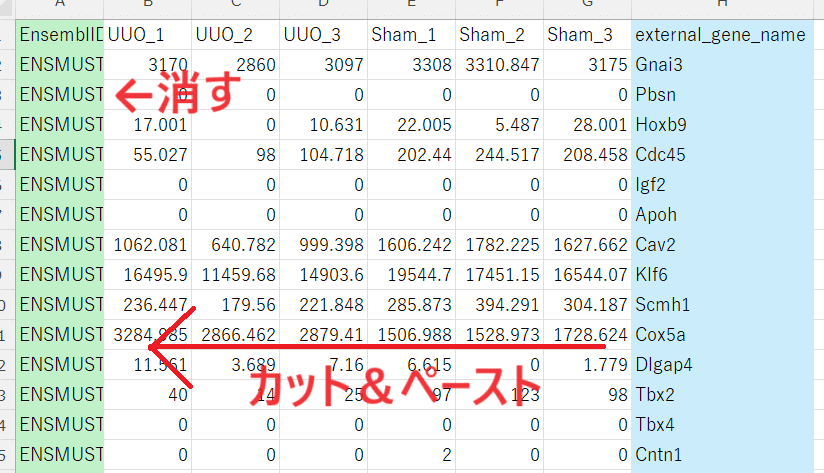
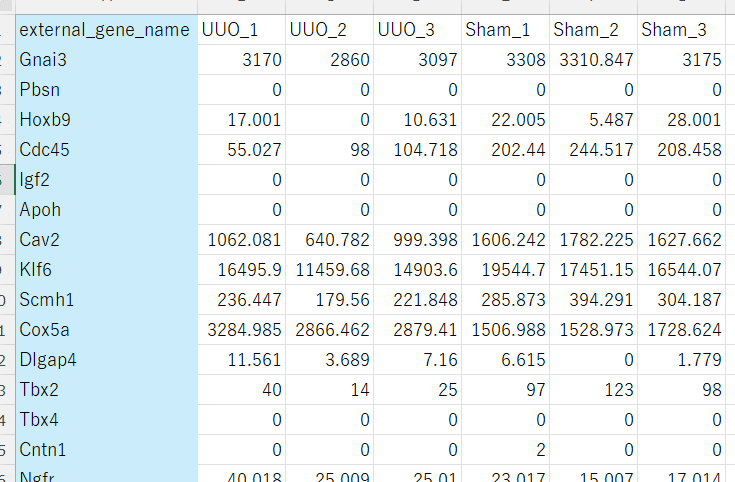
このデータをCSVファイルとして保存する。
次に、Gene symbolが「NA」もしくは「空欄」の行を削除する。
# dplyrパッケージのロード
library(dplyr)
# 列 'external_gene_name' がブランクまたはNAでない行のみを保持
merged_data_cleaned <- merged_data %>%
filter(!is.na(`external_gene_name`) & `external_gene_name` != "")続いて、Gene symbolが重複する数値列のみ合算処理を行う。
# dplyrパッケージを使用
library(dplyr)
# 数値データのみを含む列を特定
numeric_columns <- sapply(merged_data_cleaned, is.numeric)
# `external_gene_name` 列でグループ化し、数値データの列の合計値を計算
aggregated_data_sum <- merged_data_cleaned %>%
group_by(`external_gene_name`) %>%
summarise(across(where(is.numeric), sum, .names = "sum_{col}"))データを出力する
write.csv(aggregated_data_sum, "aggregated_symbol.csv", row.names = FALSE)