
【超絶初心者】SRAデータベースからRNAseq解析を初体験したメモ【RNAseqやってみたい!】

超絶初心者ですが、自分のノートPCを使ってRNAseq解析の完遂を目指しました。このnoteは、解析を再現して行うためのメモnoteです。
自分の基礎実験に並行して解析を行ったため、結構時間がかかりました(慣れれば早くなると信じています…)
長々と書いていますが、慣れたら目次の『★』がついた三項目だけ繰り返せば、カウントデータだけなら簡単に作り出せるようになっています。
以下、解析にかかった時間
・1日目:SRA toolkitの用意からsalmonを用いたマッピングまで(1サンプルのみ)
・2日目:1日目と同じ工程を全サンプルで実施
⓪PCスペック
VAIO Z
CPU 11th Gen Intel Core i7 @ 3.40 GHz
RAM 32.0 GB
Storage 1TB
①SRA toolkitの用意
下記サイトを参考にnoteを作成している。
a) SRA toolkitのインストール
下記サイトからSRA toolkitをインストールする。SRA toolkitはSRA (Sequence Read Archives) 、つまり配列に関わるデータを扱うためのツール群らしい。このツールを使うことで、NCBI SRAやDDBIなどの公共データベースから、fastqファイルやSRAファイルをダウンロードすることが可能。
下記コマンドを入力して、SRA toolkitをインストールし、解凍とパスを通す。
#SRA-toolkitのインストール
wget http://ftp-trace.ncbi.nlm.nih.gov/sra/sdk/current/sratoolkit.current-ubuntu64.tar.gz
#解凍
tar xvzf sratoolkit.current-ubuntu64.tar.gz
#解凍したファイルの移動 (verによってsratoolのファイル名が異なるため注意)
sudo mv sratoolkit.3.0.10-ubuntu64 /usr/local/src/
#パスを通す(verによってsratoolのファイル名が異なるため注意)
sudo ln -s /usr/local/src/sratoolkit.3.0.10-ubuntu64/bin/* /usr/local/bin/b) pigzのインストール(*不要かも)
*後述するgzip圧縮のが楽そうなので、これは要らないかも*
SRAファイルはとても重いので、pigz圧縮用のソフトをインストール。下記コマンドを入力。
#conda経由でpigzをインストール
conda install pigz②公共データベースからデータを探す
公共データベース(NCBI SRA)からデータを用意する。今回は「腎臓内のマクロファージ」に注目するため、下記の様にキーワードを入力して検索。

すると、下図のようにいくつかのデータが表れる。この中から、見たいデータが見つかりそうな「renal mac in WT-UUO (or sham)」に注目した。

このデータをクリックすると、下図のようなページに移動する。下の方にあるテーブルにSRRから始まるサンプル名があり、この名前を使用してubuntuのコマンド経由でSRAファイル(もしくはfastqファイル)をダウンロードする。

③SRAファイルのダウンロード
**gzip圧縮の方がワンステップで楽かも!!**
次にSRA toolkit 中の「fastq-dump」コマンドを利用して FASTQ を抽出する。
fastq-dump は、NCBIのSequence Read Archive (SRA) Toolkitの一部であるコマンドラインツール。このツールは、SRAデータベースに格納されているシーケンシングデータを、一般的に使用されるFASTQ形式に変換してダウンロードするために使用される。
a-i) fastq-dumpを使用してpigz圧縮ファイルを取得する(*不要かも)
下記のコマンドを入力することで、ペアエンドのfastqファイルをダウンロードすることが可能。ただしファイル容量がとても重いので、下記コマンドを更に施行してファイルを圧縮する。
#SRAファイル(ペアエンド)を圧縮せずに取得
fastq-dump --split-files SRR12344183
#取得したファイルをpigz形式に圧縮
pigz SRR12344183_1.fastq
pigz SRR12344183_2.fastq
a-ii) fastq-dumpを使用してgzip圧縮ファイルを取得する(★)
ちなみに、下記コマンドで直接gzip形式で圧縮ファイルを作成してもいい。こっちの方が楽かもしれない。(gzipはpigzと似たような圧縮方法。pigzの方が圧縮速度が速いらしいが、下記のコマンドのようにfastq-dumpで一括で圧縮ファイルをダウンロードする方法がない)
#SRAファイル(ペアエンド)をgzip圧縮して直接ダウンロード
fastq-dump --gzip --split-files SRR12344183
④Trim Galoreを用いてトリミング
下記サイトを参考にした。
a) Trim Galore!のインストール
まずはconda経由でTrim Galore!をインストールする。
#conda経由でTrimGalore!のインストール
conda install trim-galoreb) cutadaptおよびFastQCのインストール
次に、brew経由でcutadaptとFastQCをインストールする。これらのアプリがないと、Trim Galoreが作動できない。
brew install cutadapt
brew install FastQCc) Trim Galore!を用いたトリミング(★)
上記をインストールし、Trim Galoreのパスを通したら、実際にトリミングを行う。
まずはトリミングを行いたいファイルが存在するディレクトリに移動する。そのフォルダ内で③で取得したgzip圧縮されたペアエンドfastqファイルに対して、下記のコマンドを実行する。
*なかなかに時間がかかるので注意!!(1ペアエンドのみで30-40分かかった…)
trim_galore --paired SRR12344183_1.fastq.gz SRR12344183_2.fastq.gz実行すると、下図のようなトリミングされたファイルが得られる。
出力は入力ファイル名_val_1.fq、入力ファイル名_val_2.fqとなる。

⑤Salmonを使ったマッピング
a) Salmonのインストール
今回はSalmonを用いてマッピングを進める。
まずはSalmonをHomebrewもしくはconda経由でインストールする。今回はbrewを使用してインストールした。
#condaを用いたインストール
conda install -c bioconda -y salmon#Homebrewを用いたインストール
brew install salmonb) 公共データベースからtranscript.fastqファイルの取得
次に、salmonを使ってtranscriptのfastqファイルにindexをつけるわけだが、まずはこのfastqファイルをデータベースから取得する。今回はhumanのcDNAデータを取得する。
transcriptのfastqファイルは、EnsembleのcDNAデータを使用する。下記からアクセス可能。今回はコマンドでダウンロードを試みる。
*参考までに、下図の様に辿り着けます。下図はヒトデータの場合です!マウスとヒトの取り間違いに注意してください!!(ヒトとマウスを間違って進めてたなんて言えない…)
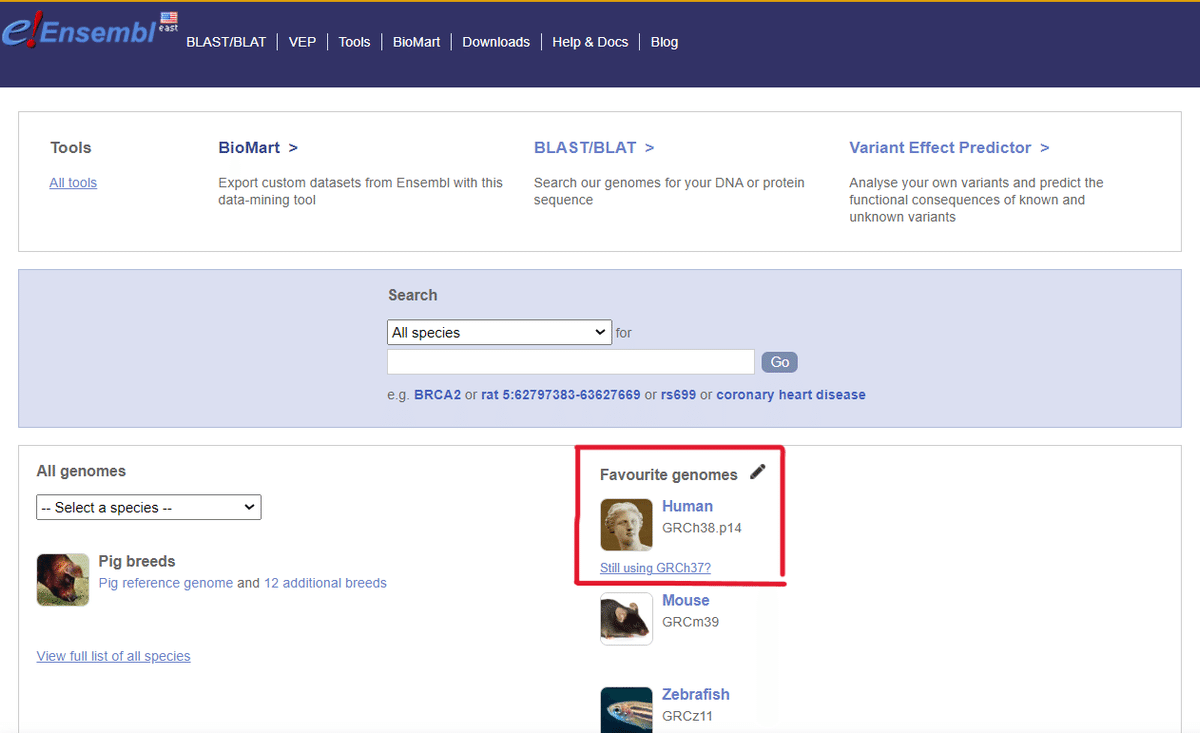
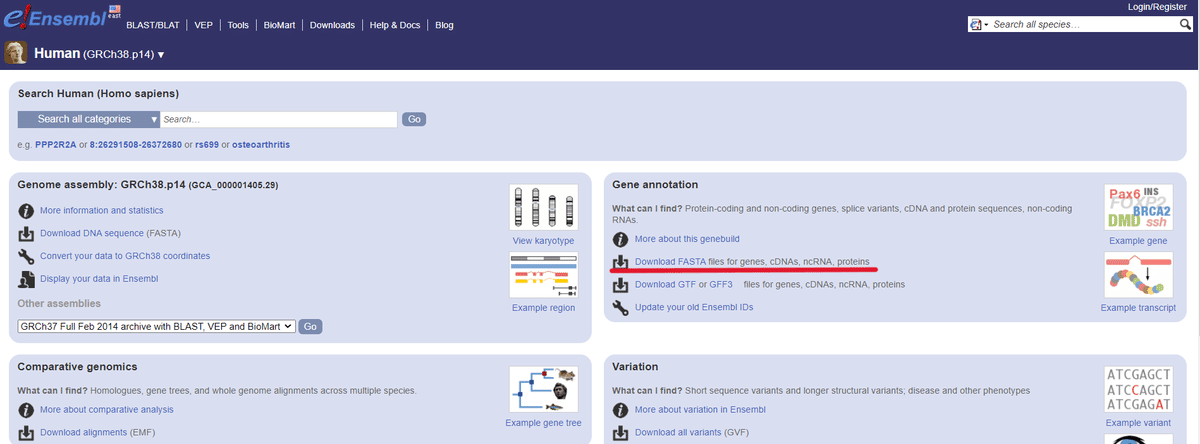


では、下記のコマンドを入力して、今回は「Mouse transcript.fastq」ファイルを取得する。
#マウスのトランスクリプトファイルをコマンドで取得
mkdir -p ~/salmon_index/salmon_mouse
cd salmon_index/salmon_mouse/
wget ftp://ftp.ensembl.org/pub/release-110/fasta/mus_musculus/cdna/Mus_musculus.GRCm39.cdna.all.fa.gz
gunzip Mus_musculus.GRCm39.cdna.all.fa.gz#ヒトサンプル用(トランスクリプトファイルの取得)
mkdir -p ~/salmon_index/salmon_human
cd salmon_index/salmon_human/
wget ftp://ftp.ensembl.org/pub/release-110/fasta/homo_sapiens/cdna/Homo_sapiens.GRCh38.cdna.all.fa.gz
gunzip Homo_sapiens.GRCh38.cdna.all.fa.gz上手くいかないときは、サイトから直接ダウンロードしてもよい。
c) Indexの作成
次にダウンロードしたtranscriptファイルにindexをつける。
下記のコマンドを実施。
#ダウンロードしたトランスクリプトファイルにindexをつける
#マウス用!
cd salmon_index/salmon_mouse/
salmon index -t Mus_musculus.GRCm39.cdna.all.fa -i Mus_musculus.GRCm39_salmon_index#ヒトサンプル用(トランスクリプトファイルにindexをつける)
cd salmon_index/salmon_human/
salmon index -t Homo_sapiens.GRCh38.cdna.all.fa -i Homo_sapiens.GRCh38_salmon_index

d) カウントの実施(定量化)(★)
まず、ここからフォルダやファイルの場所をしっかり決めないと、どれがどれだか分かりにくくなりそうなので注意。
→ 上(⑤-(c))で作成したindexフォルダと④で作成したトリミングフォルダを、同一フォルダ内に用意しておくといい。今回は、「salmon_index/salmon_mouse」の中に一緒に置いた。
フォルダを作成したら、このフォルダにディレクトリを移動。

続いて、ランを実行する。
下記のコマンドを入力して実行する。
実行後のファイルは、新しく「salmon_output」というoutputディレクトリに作成されるように指示。
**必ず出来上がるフォルダ名がサンプル毎に変わるように、下のコマンドを適宜修正すること(3か所変更する部位がある)!!フォルダ名が同じだと、過去のデータが上書きされてしまって、やり直しになる**
#ランの実施
#マウスサンプル用!
mkdir -p ~/salmon_index/salmon_mouse/salmon_output
cd ~/salmon_index/salmon_mouse/salmon_output
salmon quant -i ~/salmon_index/salmon_mouse/Mus_musculus.GRCm39_salmon_index \
-l IU \
-1 ~/salmon_index/salmon_mouse/SRR12344183_1_val_1.fq.gz \
-2 ~/salmon_index/salmon_mouse/SRR12344183_2_val_2.fq.gz \
--validateMappings \
-o ~/salmon_index/salmon_mouse/salmon_output/SRR12344183_fastq_salmon_quant#ヒトサンプル用
mkdir -p ~/salmon_index/salmon_human/salmon_output
cd ~/salmon_index/salmon_human/salmon_output
salmon quant -i ~/salmon_index/salmon_human/Homo_sapiens.GRCh38_salmon_index \
-l IU \
-1 ~/salmon_index/salmon_human/SRRXXXXXXXX_1_val_1.fq.gz \
-2 ~/salmon_index/salmon_human/SRRXXXXXXXX_2_val_2.fq.gz \
--validateMappings \
-o ~/salmon_index/salmon_human/salmon_output/SRRXXXXXXXX_fastq_salmon_quant*SalmonのlibTypeについて*
Salmonでの libType (ライブラリタイプ)の選択は、使用しているRNAシーケンシングデータの特性に基づいて行います。libType は、リードがシーケンシングされた方法と、そのリードが配列にどのように配向しているかを示します。これには、シングルエンドかペアエンドのシーケンシングか、およびリードの向きが関係します。
ライブラリタイプの決定:
シングルエンド(Single-End) vs ペアエンド(Paired-End):
シングルエンドライブラリは、各シーケンスリードが独立していることを意味します。
ペアエンドライブラリでは、リードがペアであり、一方のリードが他方の補完的な情報を持っています。
リードの配向(Orientation):
RNAシーケンシングの実験プロトコルにより、リードの向きが異なります。これは、ライブラリの構築方法に依存します。
具体的な libType オプション:
IU(Inward, Unstranded):ペアエンドライブラリで、リードが互いに向き合っている("inward")が、ストランド情報がない("unstranded")場合。
ISF(Inward, Stranded, Forward):ペアエンドライブラリで、リードが互いに向き合っていて、一方のリードが転写されたストランドと同じ方向("forward")にある場合。
ISR(Inward, Stranded, Reverse):ペアエンドライブラリで、リードが互いに向き合っていて、一方のリードが転写されたストランドの反対方向("reverse")にある場合。
SR(Single-End, Reverse):シングルエンドライブラリで、リードが転写されたストランドの反対方向にある場合。
ライブラリタイプの確認方法:シーケンシングデータを提供したラボまたはサービスプロバイダからの情報を確認します。
実験プロトコルや利用したキットの説明を参照します。
シーケンシングデータの特性を分析し、リードの向きを推測します。
ライブラリタイプは、Salmonによる解析の精度に直接影響を与えるため、正確な情報を得ることが非常に重要です。不確かな場合は、実験を実施したラボや同僚に確認することをお勧めします。また、Salmonには自動的に最適なライブラリタイプを推測する機能もありますが、可能な限り正確な情報を指定することが望ましいです。
実行後にoutputフォルダ内に、下記のように「quant.sf」ファイルができる!

⑥群間比較のためにサンプルの用意
a) すべてのサンプルで操作を繰り返す
ここまでの操作を全てのサンプルで繰り返す(目次の「★」がある項を全て)。
quant.sfファイルを作成したサンプルに関しては、SRAファイルやトリミング後のファイルは不要となるので、適宜削除して可。
ここはパワープレイ!!
b) できあがったファイルたちをフォルダにまとめる
この後のRstudioでの解析にむけて、出来上がったファイルたちをフォルダにまとめる。今回は⑤-dで出来上がったフォルダ「salmon_output」内に、下図のようにまとめた。
ここまでLinux (Ubuntu)で操作をしてきたが、ここからはRstudioを用いての解析に移る。操作をしやすくするため、この「salmon_output」フォルダをデスクトップとかドキュメントフォルダ内にコピペしておく。
⑦tximportによるデータの整理
a) Rstudioにtximportをインストールする
あまりにもRstudioに詳しくないので、ネットの情報を見様見真似で行いました。参考にしたの下記サイト。
まず、Rstudioのワーキングディレクトリをデータをまとめておいたフォルダに移動しておきます。下図の矢印(↑)のところから、「salmon_output」フォルダを選ぶ。

次に、tximportをインストールする。下記のコードをコンソールに入力し、実行する。
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install("tximport")b) tximportでquant.sfファイルを整理する
次に、tximportを使ってquant.sfファイルを整理し、この後の群間比較に使えるデータに変換する。
下記のコマンドをコンソールに入力し、実行する。
*ワーキングディレクトリ内に「SRRXXXXXX_fastq_salmon_quant」ファイルたちがあることを確認する!これらが無いとうまく走らない。
library(tximport)
library(jsonlite)
library(readr)
salmon_dirs <- list.files('.', pattern = 'fastq_salmon_quant')
salmon_dirs
salmon.files <- file.path(salmon_dirs, 'quant.sf')
names(salmon.files) <- c('UUO1', 'UUO2', 'UUO3', 'Sham1', 'Sham2', 'Sham3')
tx.exp <- tximport(salmon.files, type = "salmon", txOut = TRUE)
library(tximport)
library(jsonlite)
library(readr)
salmon_dirs <- list.files('.', pattern = 'fastq_salmon_quant')
salmon_dirs
salmon.files <- file.path(salmon_dirs, 'quant.sf')
names(salmon.files) <- c('UUO_1', 'UUO_2', 'UUO_3', 'Sham_1', 'Sham_2', 'Sham_3')
tx.exp <- tximport(salmon.files, type = "salmon", txOut = TRUE)
head(tx.exp$counts)
## UUO_1 UUO_2 UUO_3 Sham_1 Sham_2 Sham_3
## AT1G19090.1 0.000 0.000 0.000 0.000 0.000 0.000
## AT1G18320.1 12.232 34.056 27.443 10.766 15.972 12.213
## AT5G11100.1 154.000 183.000 170.000 162.000 180.000 167.000
## AT4G35335.1 455.000 509.000 514.000 399.000 492.000 467.000
## AT1G60930.1 158.000 200.000 176.000 120.000 171.000 157.000
## AT1G17000.1 0.000 0.000 0.000 0.000 0.000 0.000続いて、転写産物の名前と遺伝子名の対応表(データフレーム)を作成し、遺伝子発現量に換算する(よく理解できていない…)。
tx2gene <- data.frame(
TXNAME = rownames(tx.exp$counts),
GENEID = sapply(strsplit(rownames(tx.exp$counts), '\\.'), '[', 1)
)tx2gene データフレームの作成
データフレームの作成:
data.frame 関数を使用して、TXNAME と GENEID の2つの列を持つ新しいデータフレームを作成します。
TXNAME の割り当て:
rownames(tx.exp$counts) は、tx.exp$counts 行列(またはデータフレーム)の行名を取得します。これらの行名は通常、トランスクリプト名(たとえば、遺伝子の異なるトランスクリプトバージョンを表す)です。
GENEID の割り当て:
sapply(strsplit(rownames(tx.exp$counts), '\\.'), '[', 1) は、トランスクリプト名を . で分割し、最初の部分(通常は遺伝子IDを示す)を取得します。
ここで strsplit は文字列を分割する関数で、sapply は分割された各要素に対して関数 [', 1](最初の要素を取得する関数)を適用します。
salmon.gene.exp <- summarizeToGene(tx.exp, tx2gene)
summarizing abundance
summarizing counts
summarizing lengthデータを書き出す
csvファイルとしてデータを書き出す。
table <- as.data.frame(salmon.gene.exp$counts)
write.csv(table, file = "salmon.gene.csv", row.names = TRUE)