
Python(1) ある特定の種のQuality scoreだけ、fastqファイルから抜き出したい

背景
大学の実習において、メタゲノムや環境DNAをnanoporeで調べると、その解析結果として読み取れた全てのリードがまとまったfastq形式のファイルが出力されました。
テキストエディットのアプリを使って中身を見てみると、1つ1つのリードのデータは以下のような形で収納されています。
@93843a48-d592-4132-8ec0-1e1afa7984a4:30:257:-1:MiFish-U-F-BC13:MiFish-U-R
AGAACGATTCCATACCGTGTGAGTCGGTAAAACTCGTGCCAGTCACCGCGGTCATGATTAACCCAAGCTAACAGGGAGAGTACATAAAACGTGTTAAAGCACCATACCAAATATAAGGAATTTAACTAAGCTGTAAAAGCCATGATTAAAATAAAAATAAATGACGAAAGTGATCCCACAATAGCCGACGCACTATAGCTAAGACCCAAACTGGGATTAGATACCCCA
+
-020.,%'4323,('('%&&0.-.0011>7.,,+-++++.*))%%%%()233(%&2644:73*)+,*)+8786/3-*4,((**))59322&&)%)//+)'('*((1187**((&%#&0&%)*-&%$$$&'(211220***045:;767;869@AB2/089:6986/-120/96>;30110009---88///(()-)''&*0011611*.&&&(+00+*(&%))()%'1一番上の@以降はリード番号
2行目は塩基配列
4行目のAsciiコードで示されているのが、各塩基に対応するQuality Scoreです。
普通はFastQCなどを用いて、取った全リードに対して、Quality Scoreをチェックするそうです。
ただ実習の実験では、淡水魚の環境DNAを読んだはずなのに、メバル属(Genus Sebastes, イタリックじゃないのは許してください)の配列がバカみたいに増えてしまいました。
そこでどの過程に問題があるのか調べるため、まず持ってメバル属と判定されたリードだけfastqファイルから抜き出して、そのシーケンスに対してのみQC(クオリティチェック)したいと思ったわけです。
以上が目的です。
方法
今回使ったのはmac OSです。
考えたやり口はこうです。既に手元にあるデータとして、
先述のfastqファイル
上記のfastqファイルをBlastNをかけた結果が入っている、fasta.blastファイル
があります。そこで以下の手順でメバル属と判定されたデータのみ抜き出そうと考えました。
fasta.blastファイルから、メバル属と判定されたリードのリード番号を取得
取得したリード番号を用いて、fastqファイルから必要データを抽出
再度、クオリティチェック
1. fasta.blastファイルから、メバル属と判定されたリードのリード番号を取得
第一段階として、fasta.blastファイルから、メバル属と判定されたリードのリード番号を取得します。得られればなんでもいいのですが、私は実験でMEGANを使って結果を見ていたため、次のような方法を取りました。
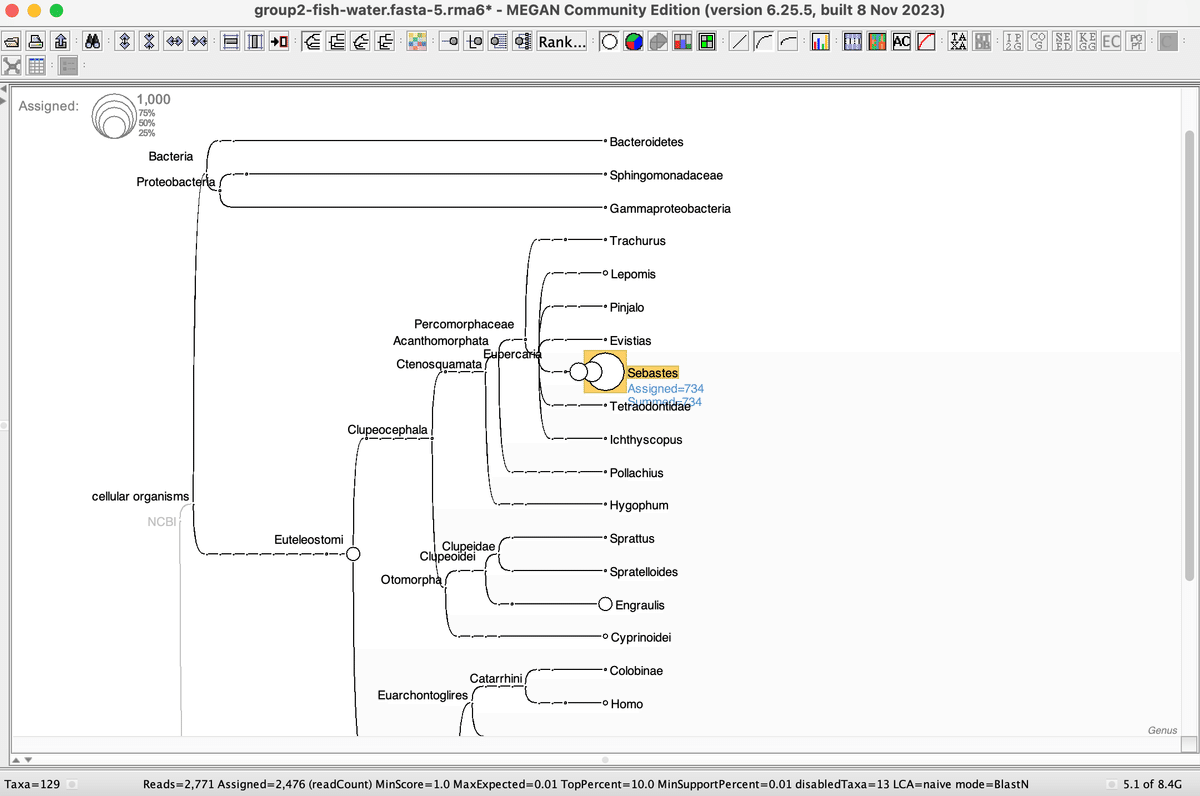
MEGANでfasta.blastファイルを開き、対象の分類群(今回はメバル属; Genus Sebastes)を選択
選択した状態で右クリック
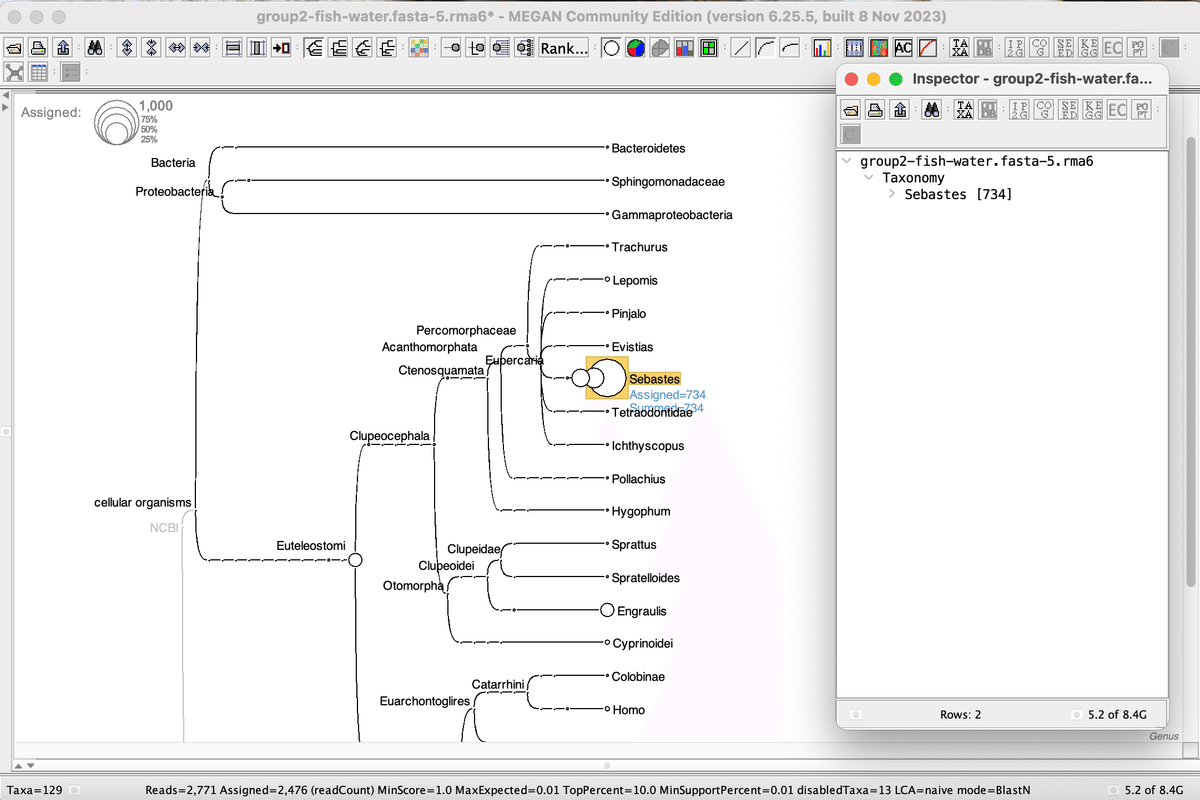
Inspectを開く
対象の分類群と判定された全リードの番号を見る
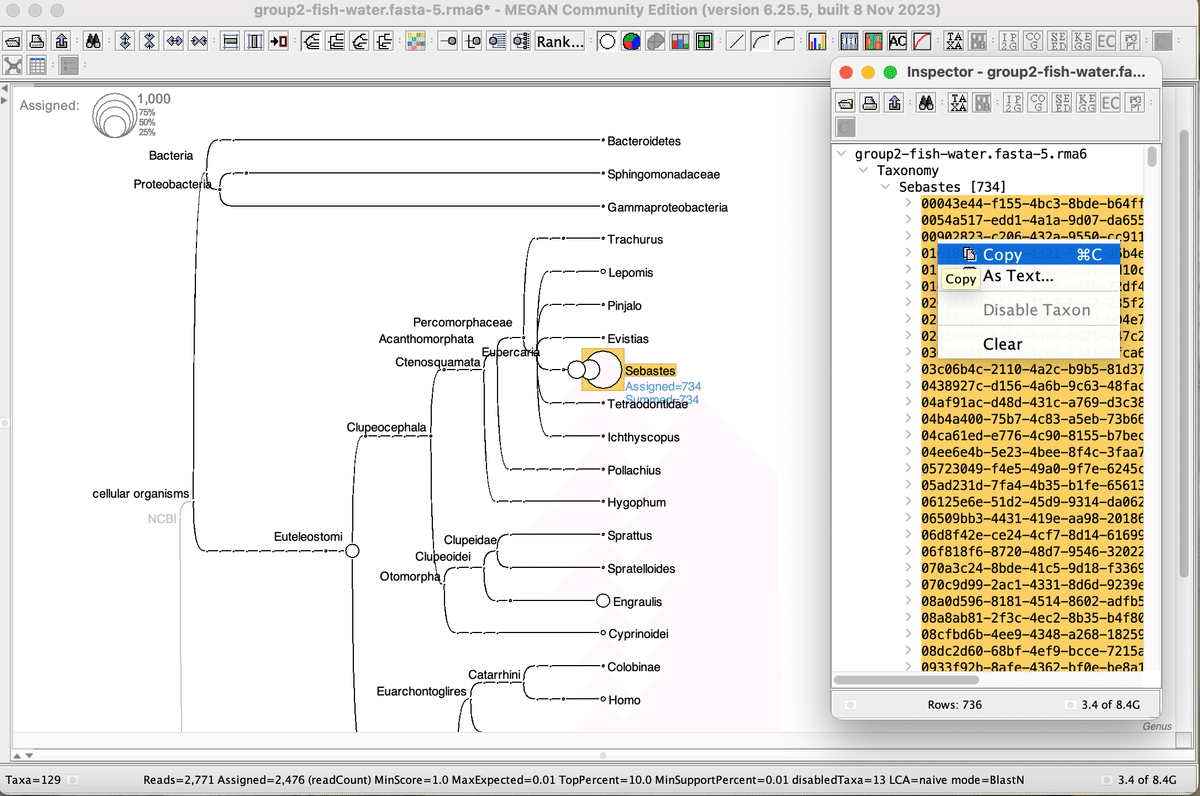
リード番号を全選択
コピー



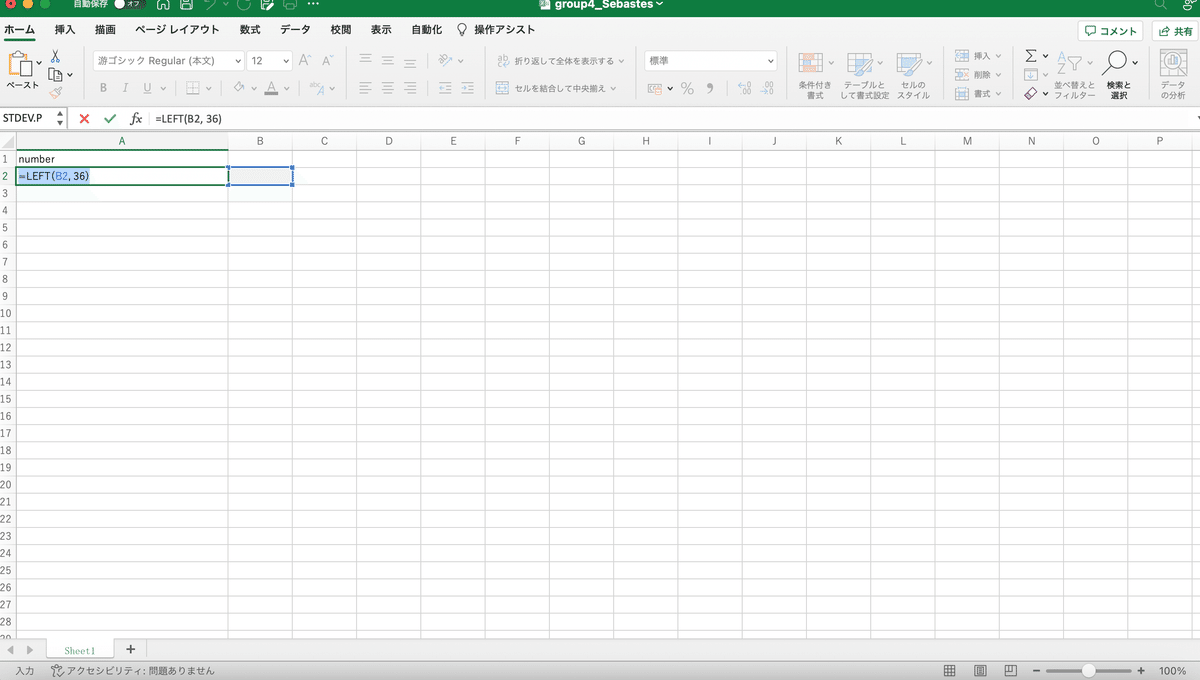
以上で得たリード番号をエクセルに貼り、LEFT関数を用いて先頭36字だけを取得することで、純粋なリード番号のリストを作成した。



これでfasta.blastファイルから、メバル属と判定されたリードのリード番号を取得の作業が終了。
MEGANを用いなくてもなんでも、リード番号がわかればいい。
2. 取得したリード番号を用いて、fastqファイルから必要データを抽出
以上で取得したリード配列を用いて、fastqファイルから必要なデータを抽出する。
今回はGoogle Colaboratory上でpythonを動かして、上記作業を行なった。
また、簡便のため、今回はデータをMy driveにアップロードして作業した。
まず以下のモジュール等の準備をする。
! pip install biopython #Biopythonパッケージのインストール
from Bio import SeqIO #bipythonからSeqIOをimport
import pandas as pd #pandasライブラリのインストール
import matplotlib.pyplot as plt #matplotライブラリのインストール次にデータを読み込む。今回は省略して、記載する
fastq_file = '/content/drive/MyDrive/ファイル名.fq'
target_numbers_file = pd.read_excel('/content/drive/MyDrive/ファイル名.xlsx') #対象種のIDをまとめたファイル以上が完了したら、データを抽出していく
extract_records=[] #抽出後のデータを入れる空のリスト
all_records = [] #比較用の全リードのデータを入れる空のリスト
with open(fastq_file, 'r') as handle: #fastqファイルを開く
for seq_record in SeqIO.parse(handle, "fastq"): #fastqファイルを読み込み、それぞれのリードデータに対して繰り返し処理をしていく
record_id = seq_record.id.split(":")[0] #リード番号の形式が一貫しているとみなして、リード番号を取得する
quality_scores = seq_record.letter_annotations["phred_quality"] #クオリティスコアを取得、詳しい文法はSeqIOの解説ページ参照
all_records.append(sum(quality_scores)/len(quality_scores)) #all_recordsというリストに、各リードのクオリティスコアの平均を入れていく
if record_id in target_numbers_file['number'].to_list(): #リード番号がメバル属のものであったら...という条件分岐
extract_records.append(sum(quality_scores)/len(quality_scores)) #extract_recordsというリストに、メバル属と判定された各リードのクオリティスコアの平均を入れていく以上のコードを実行すると、
全リードのクオリティスコア平均値が入ったall_recordsというリスト
メバル属と判定されたリードのクオリティスコア平均値が入ったextract_recordsというリスト
が手に入る
※各塩基ごとにクオリティスコアは割り当てられるため、リードごとにクオリティスコアの平均値を出している
3. クオリティチェック
最後、上記で得たデータを用いてグラフを書くことで、クオリティチェックをする
各グラフは横軸がクオリティスコア、縦軸がリード数のヒストグラムである
以下のコードで書く
#メバル属のリードに関するグラフ
plt.hist(extract_records,
bins = range(9, 24),
histtype = 'step',
density = 'True',
label="group2 Genus $\it{ Sebastes }$" + " ( n = " +str(len(all_records)) + (')'))
#全シーケンスの結果を重ね合わせる
plt.hist(all_records,
bins = range(9, 24),
histtype = 'step',
density = 'True',
label = "group2 all sequences" + " ( n = " +str(len(extract_records)) + (')'))
plt.legend(loc = 'upper center',
bbox_to_anchor = (0.5, -0.1))その結果が以下のグラフ。
メバル属のデータのクオリティが全体に対して、著しく低いわけではないことがわかる(もちろんメバル属のリード数が全体のうちの半分弱を占めている影響もあるのだろうけど)

以上のグラフデータはFastQCで言うところの、Per sequence quality scoresに相当する
参考:Biopythonパッケージに関して、参考にした日本語ページ
https://biomedicalhacks.com/2020-05-12/biopython-basic-1/
https://bi.biopapyrus.jp/python/biopython/seqio.htm
ちなみにFastQCで全リードのチェックをすると下のような感じ。

結果
ということでメバル属と全体のリードのクオリティをヒストグラムで比較することができた。
こんなことをしなくても、既にこういうことができるパッケージ等あるかもしれないので、知っている方は教えていただけると幸いです。