
老化の特徴(論文の要約)

https://doi.org/10.1016/j.cell.2013.05.039
・加齢は、生理的完全性が進行性に失われ、機能が損なわれ、死に至る危険性が増大することを特徴とする。この悪化は、がん、糖尿病、心血管障害、神経変性疾患など、ヒトの主要な病態の主要な危険因子である。特に、老化の速度は、進化の過程で保存されてきた遺伝的経路と生化学的プロセスによって、少なくともある程度コントロールされていることが発見された。本総説では、特に哺乳類の老化に重点を置いて、様々な生物における老化の共通項を示す9つの暫定的な特徴を列挙する。これらの特徴とは、ゲノムの不安定性、テロメアの消耗(減少)、エピジェネティクスの変化、プロテオスタシスの喪失、栄養センシングの乱れ、ミトコンドリアの機能不全、細胞の老化、幹細胞の枯渇、および細胞間コミュニケーションの変化などである。
【はじめに】
・ここでは、老化の細胞的・分子的特徴を同定し、分類することを試みた。一般的に老化プロセスに寄与し、老化の表現型を決定すると考えられている9つのホールマーク候補を提案する(図1)。
・各特徴は、理想的には以下の基準を満たすものでなければならない: (1)正常な老化の過程で発現すること、(2)実験的に悪化させることで老化が促進されること、(3)実験的に改善させることで正常な老化が抑制され、健康寿命が延びること。
・最後の基準は、たとえ老化の一側面に限定したとしても、達成するのが最も困難なものである。このため、老化の改善に成功した介入策によって、すべての特徴が完全に支持されているわけではない。この注意点は、老化の特徴の間には広範な相互関係があり、ある特定の特徴を実験的に改善することが、他の特徴に影響を及ぼす可能性があることを示唆している。

このスキームは、ゲノムの不安定性、テロメアの消耗、エピジェネティックな変化、プロテオスタシスの喪失、調節されていない栄養センシング、ミトコンドリア機能障害、細胞老化、幹細胞の枯渇、および細胞間コミュニケーションの変化など、このレビューに記載されている9つの特徴を列挙しています。
【ゲノムの不安定性】
・老化の共通項の一つは、生涯を通じて遺伝的損傷が蓄積することである(Moskalev et al. 2012)(図2A)。さらに、ウェルナー症候群やブルーム症候群のような数多くの早老症は、DNA損傷の蓄積の増大の結果である(Burtner and Kennedy, 2010)が、これらおよびその他の早老症症候群と正常な老化との関連性は、老化の一部の側面しか再現していないという事実のため、未解決のままである。DNAの完全性と安定性は、外因性の物理的、化学的、生物学的な要因や、DNA複製エラー、自然発生的な加水分解反応、活性酸素種(ROS)などの内因性の脅威によって絶えず脅かされている(Hoeijmakers, 2009)。外因性あるいは内因性の損傷から生じる遺伝的損傷は非常に多様であり、点突然変異、転座、染色体の増減、テロメアの短縮、ウイルスやトランスポゾンの組み込みによる遺伝子破壊などが含まれる。このような損傷を最小限に抑えるため、生物はDNA修復機構の複雑なネットワークを進化させてきた。この機構は、核DNAに加えられた損傷のほとんどに対処できるものである(Lord and Ashworth, 2012)。ゲノムの安定性システムには、テロメアの適切な長さと機能性を維持するための(これについては別のホールマークのテーマであり、以下を参照)、そして、ミトコンドリアDNA(mtDNA)の完全性を確保するための特異的なメカニズムも含まれている(Blackburn et al.) DNAのこれらの直接的な損傷に加えて、ラミノパチーとして知られる核構造の欠陥は、ゲノムの不安定性を引き起こし、早老症候群につながる可能性がある(Worman, 2012)。

(A) ゲノムの不安定性とテロメアの消耗。内因性または外因性薬剤は、単一の染色体に概略的に表されるさまざまなDNA病変を刺激することができます。このような病変は、さまざまなメカニズムによって修復できます。過度のDNA損傷または不十分なDNA修復は、老化プロセスに有利です。核DNAとミトコンドリアDNA(ここでは表されていません)の両方が加齢に伴うゲノム変化にさらされていることに注意してください。BER、塩基切除修復; HR、相同組換え; NER、ヌクレオチド切除修復; NHEJ、非相同端結合; MMR、ミスマッチ修復; ROS、活性酸素種; TLS、トランス病変合成; SAC、スピンドルアセンブリチェックポイント (Vijg, 2007)。
(B) エピジェネティックな変化。DNAのメチル化またはヒストンのアセチル化とメチル化、および他のクロマチン関連タンパク質の変化は、老化プロセスに寄与するエピジェネティックな変化を誘発する可能性があります。
①核DNA
老化したヒトやモデル生物の細胞内では、体細胞突然変異が蓄積している(Moskalev et al.,2012) 染色体異数体やコピー数の変動など、その他の形態のDNA損傷も老化と関連していることが分かっている(Faggioli et al.,2012; Forsberg et al.,2012) 大きな染色体異常によるクローン性モザイクの増加も報告されている(Jacobs et al.,2012; Laurie et al.,、2012)。これらのDNA変化の形態はすべて、必須遺伝子や転写経路に影響を及ぼす可能性があり、その結果、細胞の機能不全が生じ、アポトーシスや老化によって排除されなければ、組織や生物の恒常性が損なわれる可能性がある。このことは、DNA損傷が幹細胞の機能的能力に影 響を及ぼし、組織再生における幹細胞の役割を損なう場合に、特 に重要である(Jones and Rando, 2011; Rossi et al.,2008)(「幹細胞の枯渇」を参照)
ゲノム損傷の生涯にわたる増加と老化の関連性を示唆する原因証拠は、マウスやヒトを用いた研究から得られており、DNA修復機構の欠損がマウスの老化を促進し、ウェルナー症候群、ブルーム症候群、色素性乾皮症、トリコチオジストロフィー、コケイン症候群、セッケル症候群など、ヒトのいくつかの早老症候群の根底にあることが示されている(Gregg et al.,2012; Hoeijmakers,2009; Murga et al.,2009) さらに、染色体の正確な分離を確実にする有糸分裂チェックポイントの構成要素であるBubR1を過剰発現させたトランスジェニックマウスは、異数性とがんに対する防御が強化され、健康寿命も延びた(Baker et al.,2013)。 後者の発見は、核DNA修復機構の人為的強化が老化を遅らせる可能性があるという実験的証拠を示している。
②ミトコンドリアDNA
老化したミトコンドリアDNAの変異や欠失も老化に関与している可能性がある(Park and Larsson, 2011)。ミトコンドリアの酸化的微小環境、ミトコンドリアDNAには保護ヒストンが存在しないこと、核DNAの修復機構に比べてミトコンドリアDNAの修復機構の効率が限られていることなどから、ミトコンドリアDNAは老化に伴う体細胞変異の主要な標的であると考えられてきた(Linnane et al.,1989) 老化におけるmtDNA変異の因果関係については、ミトコンドリアゲノムの多様性により、同一細胞内に変異型ゲノムと野生型ゲノムが共存する、いわゆる "ヘテロプラスミー "と呼ばれる現象が起こるため、論争が続いてきた。しかし、単一細胞の解析から、mtDNA変異の全体的なレベルは低いにもかかわらず、個々の老化細胞の変異負荷は大きくなり、1つの変異ゲノムが優勢なホモプラスミーの状態になる可能性があることが明らかになった(Khrapko et al.,1999) 興味深いことに、これまでの予想に反して、成体細胞や老化細胞におけるmtDNA突然変異のほとんどは、酸化的損傷によってではなく、むしろ生後早期の複製エラーによって引き起こされるようである。これらの変異は多クローン性拡大を起こし、異なる組織で呼吸鎖機能不全を引き起こす可能性がある(Ameur et al.,2011) mtDNAの複製を阻害する抗レトロウイルス薬で治療を受けているHIV感染患者における老化の加速に関する研究は、生後早期に生じたmtDNA変異のクローン性拡大という概念を支持している(Payne et al.,2011)
mtDNAの損傷が老化や加齢関連疾患にとって重要であるかもしれないという最初の証拠は、部分的に老化を表現型コピーするmtDNA変異によって引き起こされるヒトの多系統疾患が同定されたことに由来する(Wallace, 2005)。さらに、ミトコンドリアDNAポリメラーゼγを欠損したマウスの研究から、その原因となる証拠が得られている。これらの変異マウスは、mtDNAにおけるランダムな点突然変異や欠失の蓄積に関連して、早期老化や寿命短縮の様相を示す(Kujoth et al., 2005;Trifunovic et al.,2004;Vermulst et al.,2008)。これらのマウスの細胞はミトコンドリア機能に障害を示すが、予想外なことに、これは活性酸素(ROS)産生の増加を伴わない(Edgar et al., 2009;Hiona et al., 2010)。さらに、これらのプロジェロイド(早老性)マウスの幹細胞は、特にmtDNA変異の蓄積に敏感である(Ahlqvist et al., 2012)(「幹細胞の枯渇」参照)。mtDNA変異の負荷を減少させる遺伝子操作が寿命延長につながるかどうかを判断するには、今後の研究が必要である。
③核構造
核ラミナ(ほとんどの真核細胞の核膜の内側に存在する、厚さ約30–100 nmの網状の構造)の欠陥もまた、ゲノムの不安定性を引き起こす可能性がある(Dechat et al.,2008) 核ラミンは核ラミナの主要な構成要素であり、クロマチンやゲノムの安定性を制御するタンパク質複合体を繋ぎとめる足場を提供することで、ゲノムの維持に関与している(Gonzalez-Suarez et al.,2009; Liu et al.,2005) 核ラミナは、この構造を構成するタンパク質をコードする遺伝子の変異や、核ラミナの成熟や動態に影響を及ぼす因子が、Hutchinson-Gilford症候群やNéstor-Guillermo progeria症候群(それぞれHGPSとNGPS)のような老化促進症候群を引き起こすことが発見された後、老化研究者の注目を集めるようになった(Cabanillas et al.,2011; De Sandre-Giovannoli et al.,2003; Eriksson et al.,2003) 核ラミナの変化や、プロジェリンと呼ばれる異常なプレラミンAアイソフォームの産生も、ヒトの正常な老化の過程で検出されている(Ragnauth et al.,2010;Scaffidi and Misteli,2006)。テロメアの機能不全もまた、正常なヒト線維芽細胞において、長期間のin vitro培養によりプロジェリン産生を促進することから、正常な老化におけるテロメアの維持とプロジェリン発現との密接な関係が示唆される(Cao et al.,2011) このようなA型ラミンの老化に伴う変化に加えて、細胞の老化に伴ってラミンB1レベルが低下することから、このプロセスのバイオマーカーとしての有用性が指摘されている(Freund et al.,、2012;Shimi et al., 2011)。
動物モデルや細胞モデルによって、HGPS(ハッチンソン・ギルフォード・プロジェリア症候群)に特徴的な核ラミナの異常によって惹起されるストレス経路の同定が容易になった。これらの経路には、p53の活性化(Varela et al., 2005)、体性軸の調節解除(Mariño et al.,2010)、成体幹細胞の減少(Espada et al., 2008;Scaffidi and Misteli、2008)などが含まれる。早老における核ラミナ異常の因果関係は、HGPSモデルマウスにおいて、プレラミンAやプロジェリンレベルを低下させることで、早老症の特徴の発現が遅延し、寿命が延長するという観察によって支持されている。これは、アンチセンスオリゴヌクレオチド、ファルネシルトランスフェラーゼ阻害剤、またはスタチンとアミノビスホスホネートの組み合わせの全身注射によって達成できる(Osorio et al., 2011;Varela et al., 2008;Yang et al., 2006)。ホルモン治療やNF-κBシグナルの阻害による体性軸の回復も、これらの早老症マウスの寿命を延長させる(Mariño et al.、2010;Osorio et al., 2012)。さらに、HGPS患者由来の人工多能性幹細胞(iPSCs)のLMNA変異を修正する相同組換えベースの戦略が開発され、将来の細胞治療への道が開かれた(Liu et al.,2011b)。核構造の強化が正常な老化を遅らせるという考えを検証するためには、さらなる研究が必要である。
【テロメアの消耗(減少)】
加齢に伴うDNA損傷の蓄積は、ゲノムにほぼランダムに影響するように見えるが、テロメアなど、加齢による劣化の影響を特に受けやすい染色体領域もある(Blackburn et al.、2006)(図2A)。複製型DNAポリメラーゼは直鎖DNA分子の末端を完全に複製する能力を持たないが、この機能はテロメラーゼとして知られる特殊なDNAポリメラーゼが独自に持つ。しかし、ほとんどの哺乳類の体細胞はテロメラーゼを発現していないため、染色体末端からテロメア保護配列が徐々に、そして累積的に失われていく。テロメアの枯渇は、ある種の体外培養細胞の限られた増殖能力、いわゆる複製老化、あるいはヘイフリック限界(Hayflick and Moorhead, 1961; Olovnikov, 1996)を説明する。実際、テロメラーゼの異所性発現は、発癌性形質転換を起こすことなく、そうでなければ死に至る細胞に不死性を与えるのに十分である(Bodnarら、1998)。重要なことに、テロメアの短縮はヒトでもマウスでも正常な老化の過程で観察される(Blasco, 2007a)。
テロメアはシェルタリンとして知られる特徴的な多タンパク質複合体によって結合されている(Palm and de Lange, 2008)。この複合体の主な機能は、DNA修復タンパク質がテロメアにアクセスするのを防ぐことである。そうでなければ、テロメアはDNA切断として「修復」され、染色体の融合につながってしまう。DNA修復が制限されるため、テロメアのDNA損傷は顕著に持続し、老化やアポトーシスを誘導するのに非常に効果的である(Fumagalli et al.)
ヒトにおけるテロメラーゼ欠損は、肺線維症、先天性角化不全症、再生不良性貧血など、様々な組織の再生能力の喪失を伴う疾患の早期発症と関連している(Armanios and Blackburn, 2012)。テロメアのアンキャッピングや染色体融合が多発するのも、シェルテリン成分の欠損が原因である(Palm and de Lange, 2008)。シェルテリンの突然変異は、再生不良性貧血や先天性角化異常症のいくつかの症例で見つかっている(Savageら、2008;Walneら、2008;Zhongら、2011)。シェルタリン成分の様々な機能喪失モデルは、組織の再生能力の急速な低下と老化の促進を特徴としており、この現象は正常な長さのテロメアが存在する場合でも起こる(Martínez and Blasco, 2010)。
遺伝子改変動物モデルでは、テロメアの消失、細胞の老化、生物の老化の間に因果関係があることが立証されている。テロメアが短くなったり長くなったりしたマウスは、それぞれ寿命が短くなったり長くなったりする(Armanios et al., 2009; Blasco et al., 1997; Herrera et al., 1999; Rudolph et al., 1999; Tomás-Loba et al., 2008)。最近の証拠は、テロメラーゼの活性化によって老化が回復することも示している。特に、テロメラーゼ欠損マウスの早期老化は、これらの老化マウスでテロメラーゼを遺伝的に再活性化すると元に戻る(Jaskelioff et al., 2011)。さらに、野生型成体マウスにテロメラーゼをウイルスで全身導入すると、がんの発生率を増加させることなく、正常な生理的老化を遅らせることができる(Bernardes de Jesus et al.) ヒトでは、最近のメタアナリシスで、短いテロメアと死亡リスクとの間に強い関係があることが、特に若い年齢で支持されている(Boonekampら、2013)。
【エピジェネティックな変化】
様々なエピジェネティックな変化は、生涯を通じてすべての細胞や組織に影響を及ぼす(Talens et al. エピジェネティックな変化には、DNAメチル化パターンの変化、ヒストンの翻訳後修飾、クロマチンリモデリングが含まれる。ヒストンH4K16アセチル化、H4K20トリメチル化、H3K4トリメチル化の増加、およびH3K9メチル化、H3K27トリメチル化の減少は、加齢に伴うエピジェネティックマークを構成する(Fraga and Esteller, 2007; Han and Brunet, 2012)。エピジェネティックパターンの生成と維持を保証する複数の酵素系には、DNAメチルトランスフェラーゼ、ヒストンアセチラーゼ、脱アセチラーゼ、メチラーゼ、脱メチラーゼのほか、クロマチンリモデリングに関与するタンパク質複合体が含まれる。
①ヒストン修飾
ヒストンのメチル化は、無脊椎動物の老化の特徴の条件を満たしている。ヒストンメチル化複合体の構成要素(H3K4とH3K27)を欠失させると、線虫とハエではそれぞれ寿命が延びる(Greer et al.) さらに、ミミズにおける(H3K27に対する)ヒストン脱メチル化酵素の阻害は、インスリン/IGF-1シグナル伝達経路のような重要な長寿経路の構成要素を標的とすることにより、寿命を延ばす可能性がある(Jin et al.) ヒストン修飾酵素の操作が、DNA修復やゲノムの安定性に影響を与えることによって、あるいは核外の代謝経路やシグナル伝達経路に影響を与える転写変化を通して、純粋にエピジェネティックなメカニズムによって老化に影響を与えるかどうかは、まだ明らかではない。
NAD依存性タンパク質脱アセチル化酵素およびADPリボシルトランスフェラーゼのサーチュインファミリーのメンバーは、潜在的な老化防止因子として広く研究されてきた。老化との関連でこのタンパク質ファミリーに関心が集まったのは、酵母、ハエ、ミミズを用いた一連の研究からであり、これらの生物のSir2と名付けられた単一のサーチュイン遺伝子が顕著な長寿活性を持つことが報告された(Guarente, 2011)。Sir2の過剰発現は、サッカロミセス・セレビシエ(Kaeberlein et al., 1999)において複製寿命を延長することが初めて示され、その後の報告では、ミミズ(sir-2.1)とハエ(dSir2)のオルソログの発現を増強することで、無脊椎動物の両モデル系において寿命を延長できることが示された(Rogina and Helfand, 2004; Tissenbaum and Guarente, 2001)。しかしながら、これらの知見は最近になって疑問視されるようになった。ミミズとハエの研究で当初観察された寿命延長は、ほとんどが遺伝的背景の違いによるものであり、それぞれsir-2.1やdSir2の過剰発現によるものではなかったという報告である(Burnett et al., 2011)。実際、注意深く再評価を行った結果、線虫ではsir-2.1の過剰発現は、ささやかな寿命延長しかもたらさないことがわかった(Viswanathan and Guarente, 2011)。
哺乳類に関しては、7つの哺乳類サーチュインパラログのうちいくつかは、マウスにおいて老化の様々な側面を改善することができる(Houtkooper et al.) 特に、無脊椎動物のSir2に最も近いホモログである哺乳類SIRT1のトランスジェニック過剰発現は、加齢に伴う健康状態を改善するが、寿命は延長しない(Herranz et al.) SIRT1の有益な効果に関与するメカニズムは複雑で、ゲノムの安定性の向上(Oberdoerfferら、2008;Wangら、2008)や代謝効率の向上(Nogueirasら、2012)など、相互に関連している(「調節されない栄養素感知」参照)。ヒストンH3K9脱アセチル化を通じて、ゲノムの安定性、NF-κBシグナル伝達、グルコースホメオスタシスを制御するSIRT6については、哺乳類におけるサーチュインを介したプロロンジェビティの役割について、より説得力のある証拠が得られている(Kanfiら、2010;Kawaharaら、2009;Zhongら、2010)。一方、SIRT6を過剰発現した雄性トランスジェニックマウスは対照動物よりも寿命が長く、血清IGF-1やIGF-1シグナル伝達の他の指標も減少している(Kanfi et al. 興味深いことに、ミトコンドリアに位置するサーチュインSIRT3は、食事制限(DR)の長寿における有益な効果の一部を媒介することが報告されているが、その効果はヒストン修飾によるものではなく、むしろミトコンドリアタンパク質の脱アセチル化によるものである(Someya et al.) ごく最近、SIRT3を過剰発現させると、老化した造血幹細胞の再生能力が向上することが報告された(Brown et al.) したがって、哺乳類では、サーチュインファミリーの少なくとも3つのメンバー-SIRT1、SIRT3、SIRT6-が健康な老化に寄与している。
②DNAメチル化
DNAメチル化と老化の関係は複雑である。初期の研究では、加齢に伴うグローバルなメチル化低下が報告されていたが、その後の解析で、様々な癌抑制遺伝子やポリコーム標的遺伝子に対応する遺伝子座を含むいくつかの遺伝子座が、加齢に伴って実際にメチル化亢進することが明らかになった(Maegawa et al.) プロジェロイド症候群の患者やマウスの細胞は、DNAメチル化パターンとヒストン修飾を示し、それらは正常な加齢で見られるものとほぼ同じである(Osorio et al.) 生涯を通じて蓄積されたこれらのエピジェネテ ィックな欠陥やエピミューテーションは全て、幹細胞の挙動や機能性 に特異的に影響を及ぼす可能性がある(Pollina and Brunet, 2011)(「幹細胞の枯渇」参照)。とはいえ、DNAメチル化のパターンを変化させることで生物の寿命を延ばすことができるという直接的な実験的実証は、今のところなされていない。
③クロマチンリモデリング
DNA修飾酵素やヒストン修飾酵素は、ヘテロクロマチンタンパク質1α(HP1α)などの主要な染色体タンパク質や、ポリコームグループタンパク質やNuRD複合体などのクロマチンリモデリング因子と協調して働くが、そのレベルは正常細胞でも病的老化細胞でも低下している(Pegoraro et al.) 上述したヒストンやDNAメチル化におけるエピジェネティック修飾とともに、これらのエピジェネティック因子の変化は、老化の特徴であるヘテロクロマチンの全体的な減少や再分布といったクロマチン構造の変化を決定する(Oberdoerffer and Sinclair, 2007; Tsurumi and Li, 2012)。加齢におけるこれらのクロマチン変化の因果関係は、HP1αの機能喪失変異を持つハエは寿命が短くなる一方で、このヘテロクロマチンタンパク質の過剰発現はハエの寿命を延ばし、老齢に特徴的な筋肉の衰えを遅らせるという発見によって支持されている(Larson et al.)。
老化におけるエピジェネティクスを介したクロマチン変化の機能的関連性を支持するものとして、反復DNAドメインにおけるヘテロクロマチン形成と染色体の安定性との間に注目すべき関係がある。特に、ペリセントリック領域でのヘテロクロマチン形成には、HP1α結合と同様に、ヒストンH3K9とH4K20のトリメチル化が必要であり、染色体の安定性に重要である(Schotta et al.) 哺乳類のテロメア反復配列もまた、これらのクロマチン修飾に富んでおり、染色体末端がヘテロクロマチンドメインに集められていることを示している(Blasco, 2007b; Gonzalo et al.) サブテロメア領域もまた、H3K9とH4K20のトリメチル化、HP1α結合、DNAハイパーメチル化など、構成的ヘテロクロマチンの特徴を示す。このように、エピジェネティックな変化は、老化の特徴の一つであるテロメアの長さの制御に直接影響を及ぼす可能性がある。
④転写の変化
加齢は、転写ノイズの増加(Bahar et al., 2006)と、多くのmRNAの異常な産生と成熟に関連している(Harries et al.) いくつかの生物種の若い組織と老齢組織のマイクロアレイによる比較から、炎症、ミトコンドリア、リソソーム分解経路の主要な構成要素をコードする遺伝子において、加齢に伴う転写変化が同定されている(de Magalhães et al.) このような加齢に伴う転写シグネチャーは、ノンコーディングRNAにも影響を及ぼし、その中には、加齢プロセスに関連し、長寿ネットワークの構成要素を標的としたり、幹細胞の挙動を制御したりすることで寿命に影響を及ぼす一群のmiRNA(gero-miR)も含まれる(Boulias and Horvitz, 2012; Toledano et al.) 機能獲得および機能喪失の研究により、ショウジョウバエや線虫において、いくつかのmiRNAが長寿を調節する能力があることが確認されている(Liu et al.、2012;Shen et al.、2012;Smith-Vikos and Slack、2012)。
⑤エピジェネティックな変化の回復
DNAの突然変異とは異なり、エピジェネティックな変化は少なくとも理論的には可逆的であるため、新規の抗加齢治療をデザインする機会を提供する(Freije and López-Otín, 2012; Rando and Chang, 2012)。ヒストン脱アセチル化酵素阻害剤の投与により生理的なH4アセチル化が回復すると、加齢に伴うマウスの記憶障害が回避されることから(Peleg et al. また、ヒストンアセチルトランスフェラーゼの阻害剤は、プロジェロイドマウスの早期老化の表現型を改善し、寿命を延長させる(Krishnanら、2011年)。さらに、最近発見された線虫における長寿の世代間エピジェネティック遺伝は、両親の特定のクロマチン修飾を操作することで、子孫に長寿のエピジェネティック記憶を誘導できることを示唆している(Greer et al., 2011)。ヒストンアセチルトランスフェラーゼ阻害剤と同様の概念で、ヒストン脱アセチラーゼ活性化剤も長寿を促進する可能性が考えられる。レスベラトロールは老化との関連で広く研究されており、その複数の作用機序の中には、SIRT1活性のアップレギュレーションや、エネルギー障害に関連する他の作用がある(「ミトコンドリア機能障害」参照)。
【プロテオスタシスの喪失】
加齢や、加齢に関連した疾患のいくつかは、タンパク質の恒常性(ホメオスタシス)あるいはプロテオスタシスの障害と関連している(Powers et al.) 全ての細胞は、プロテオームの安定性と機能性を維持するために、様々な品質管理機構を利用している。プロテオスタシスには、正しく折り畳まれたタンパク質(特に熱ショックタンパク質ファミリー)の安定化機構と、プロテアソームやリソソームによるタンパク質の分解機構が関与している(Hartl et al. さらに、MOAG-4のように、分子シャペロンやプロテアーゼとは異なる別の経路で作用する、加齢に伴うタンパク質毒性を制御する因子も存在する(van Ham et al.) これらのシステムはすべて、協調してミスフォールドしたポリペプチドの構造を修復したり、あるいは完全に除去して分解したりするように機能するため、傷ついた成分の蓄積を防ぎ、細胞内タンパク質の継続的な更新を保証する。従って、プロテオスタシスは加齢とともに変化することが、多くの研究で証明されている(Koga et al.) さらに、アンフォールディングタンパク質、ミスフォールディングタンパク質、凝集タンパク質の慢性的な発現は、アルツハイマー病、パーキンソン病、白内障など、加齢に関連した病態の発症に寄与している(Powersら、2009)。

①シャペロンを介したタンパク質のフォールディングと安定性
ストレスによって誘導される細胞質およびオルガネラ特異的シャペロンの合成は、加齢によって著しく損なわれる(Calderwood et al.) 多くの動物モデルが、シャペロンの減少が長寿に及ぼす原因的影響を支持している。特に、シャペロンを過剰発現させたトランスジェニックワームやハエは長寿である(Morrow et al.) また、熱ショックファミリーのコシャペロンを欠損させた変異マウスは老化を促進する表現型を示すが、長寿マウス系統ではいくつかの熱ショックタンパク質の顕著な発現上昇が見られる(Min et al., 2008; Swindell et al.) さらに、熱ショック応答のマスターレギュレーターである転写因子HSF-1の活性化は線虫の長寿と耐熱性を増加させ(Chiang et al. 哺乳動物細胞では、SIRT1によるHSF-1の脱アセチル化は、Hsp70などの熱ショック遺伝子の転写活性化を増強し、一方、SIRT1のダウンレギュレーションは熱ショック応答を減弱させる(Westerheide et al., 2009)。
プロテオスタシスを維持または増強するためのいくつかのアプローチは、シャペロンを介するタンパク質のフォールディングと安定性を活性化することを目的としている。熱ショックタンパク質Hsp72の薬理学的誘導は、筋ジストロフィーモデルマウスにおいて筋機能を維持し、ジストロフィー病態の進行を遅らせる(Gehrig et al., 2012)。低分子はまた、薬理学的シャペロンとして、損傷タンパク質のリフォールディングを確実にし、モデル生物における加齢に関連した表現型を改善するために用いられるかもしれない(Calamini et al.)
②タンパク質分解システム
タンパク質の品質管理に関与する2つの主要なタンパク質分解系、すなわちオートファジー-リソソーム系とユビキチン-プロテアソーム系の活性は加齢とともに低下し(Rubinsztein et al.
オートファジーに関しては、シャペロンを介するオートファジー受容体LAMP2aを余分にコピーしたトランスジェニックマウスは、加齢に伴うオートファジー活性の低下を経験せず、加齢とともに肝機能が改善する(Zhang and Cuervo, 2008)。マクロオートファジー(シャペロンが介在するオートファジーとは異なる別のタイプのオートファジー)の化学的誘導剤を用いた介入は、mTOR阻害剤であるラパマイシンを常時または断続的に投与すると中年マウスの寿命が延びるという発見以来、並々ならぬ関心を呼んでいる(Blagosklonny, 2011; Harrison et al.) 注目すべきは、ラパマイシンがマウスの老化を多面的に遅らせることである(Wilkinsonら、2012年)。ラパマイシンの寿命延長効果は、酵母、線虫、ハエにおけるオートファジーの誘導に厳密に依存している(Bjedovら、2010;Rubinszteinら、2011)。しかしながら、哺乳類の老化に対するラパマイシンの効果については、同様の証拠はまだ存在せず、タンパク質合成に関与するリボソームS6プロテインキナーゼ1(S6K1)の阻害(Selman et al. ラパマイシンとは対照的に、免疫抑制の副作用がない別のマクロオートファジー誘導物質であるスペルミジンもまた、オートファジーの誘導を介して酵母、ハエ、ミミズにおいて長寿を促進する(Eisenberg et al.) 同様に、スペルミジンを含むポリアミン製剤による栄養補給や、ポリアミン産生腸内細菌叢の供給は、マウスの寿命を延ばす(Matsumoto et al.) ω-6系多価不飽和脂肪酸を食事で補給すると、オートファジーの活性化によって線虫の寿命も延びる(O'Rourke et al.)。
プロテアソームとの関連では、EGFシグナルの活性化がユビキチン・プロテアソーム系の様々な成分の発現を増加させることにより、線虫の寿命を延長させる(Liu et al.) 同様に、脱ユビキチラーゼ阻害剤やプロテアソーム活性化剤によるプロテアソーム活性の増強は、ヒト培養細胞における有毒タンパク質のクリアランスを促進し(Lee et al. さらに、FOXO転写因子DAF-16によるプロテアソームサブユニットRPN-6の発現増加は、線虫においてタンパク質毒性ストレス耐性を付与し、寿命を延ばす(Vilchez et al.)。
【調節不能な栄養感知】
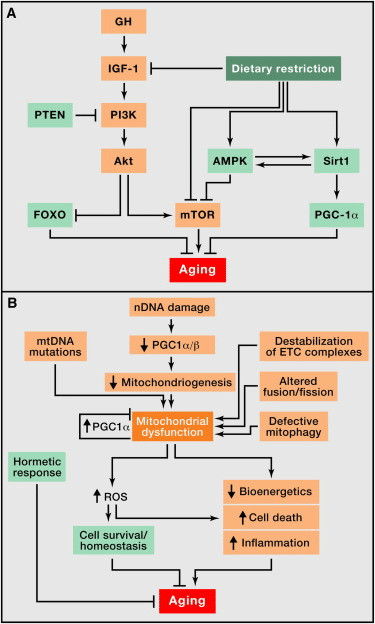
(A) 規制緩和された栄養センシング。成長ホルモン(GH)とインスリン/インスリン成長因子1(IGF-1)シグナル伝達経路と、食事制限と老化との関係を含む体質栄養軸の概要。老化を促進する分子はオレンジ色で示され、アンチエイジング特性を持つ分子は薄緑色で示されています。
(B) ミトコンドリア機能障害。ミトコンドリア機能は、老化に関連するmtDNA変異、ミトコンドリア形成の減少、電子輸送鎖(ETC)複合体の不安定化、ミトコンドリアダイナミクスの変化、またはミトファジーによる品質管理の欠陥によって乱されます。ストレス信号と欠陥のあるミトコンドリア機能がROSを生成し、一定の閾値を下回ると、生存シグナルを誘導して細胞の恒常性を回復しますが、より高いまたは継続的なレベルでは、老化に寄与する可能性があります。同様に、軽度のミトコンドリア損傷は、適応補償プロセスを引き起こすホルメティック反応(ミトホルメシス)を誘発する可能性があります。
哺乳類の体性筋軸は、下垂体前葉で産生される成長ホルモン(GH)と、その二次メディエーターであるインスリン様成長因子1(IGF-1)から構成され、GHに応答して多くの細胞タイプ、特に肝細胞で産生される。IGF-1の細胞内シグナル伝達経路は、グルコースの存在を細胞に知らせるインスリンによって誘発される経路と同じである。このため、IGF-1とインスリンのシグナル伝達は、「インスリン・IGF-1シグナル伝達」(IIS)経路として知られている。驚くべきことに、IIS経路は進化の過程で最も保存されている老化制御経路であり、その複数の標的の中には、転写因子のFOXOファミリーやmTOR複合体も含まれている(Barzilaiら、2012;Fontanaら、2010;Kenyon、2010)。GH、IGF-1受容体、インスリン受容体、あるいはAKT、mTOR、FOXOのような下流の細胞内エフェクターの機能を低下させる遺伝子多型や突然変異は、ヒトでもモデル生物でも長寿と関連しており、栄養および生体エネルギー経路が長寿に大きな影響を与えていることをさらに示している(Barzilaiら、2012;Fontanaら、2010;Kenyon、2010)(図4A)。
老化の特徴として、調節された栄養センシングの関連性と一致して、食事制限(DR)は、ヒト以外の霊長類を含む、調査されたすべての真核生物種において、寿命または健康寿命を延ばす(Colmanら、2009;Fontanaら、2010;Mattisonら、2012)。
①インスリンとIGF-1のシグナル伝達経路
IIS経路の様々なレベルでのシグナル伝達強度を減弱させる複数の遺伝子操作により、ミミズ、ハエ、マウスの寿命は一貫して延長している(Fontana et al.) 遺伝学的解析から、この経路がミミズやハエの寿命に対するDRの有益な効果の一部を仲介していることが示された(Fontana et al.) IIS経路の下流エフェクターの中で、ミミズやハエの長寿に最も関係しているのは、転写因子FOXOである(Kenyon et al.) マウスには4人のFOXOメンバーがいるが、それらの過剰発現が長寿に及ぼす影響や、IISの減少による健康寿命の延伸を媒介する役割については、まだ明らかにされていない。マウスのFOXO1はDRの腫瘍抑制効果に必要である(Yamaza et al. 最近では、腫瘍抑制因子PTENの投与量が増加したマウスが、IIS経路の全般的なダウンモジュレーションを示し、ミトコンドリアの酸化代謝の改善や褐色脂肪組織の活性の亢進に関連したエネルギー消費の増加を示すことが報告されている(Garcia-Caoら、2012;Ortega-Molinaら、2012)。IIS活性を低下させた他のマウスモデルと同様に、Pten過剰発現マウスやPI3K低形成マウスは長寿を示す(Foukasら、2013;Ortega-Molinaら、2012)。
逆説的ではあるが、GHとIGF-1のレベルは正常な加齢においても、早老モデルマウスにおいても低下する(Schumacherら、2008年)。このように、IISの低下は、生理的加齢と加速加齢の両方に共通する特徴であり、一方、IISが構成的に低下すると長寿になる。このような一見矛盾する観察結果は、IISの低下調節が、全身的なダメージの中で細胞の増殖と代謝を最小限に抑えることを目的とした防御反応を反映しているという統一的なモデルで説明することができる(Garinis et al.) この考え方によれば、IISが恒常的に低下している生物は、細胞増殖と代謝の速度が低く、したがって細胞損傷の速度も低いため、より長く生き延びることができる。これと同様に、生理的あるいは病的に老化した生物は、寿命を延ばそうとしてIISを低下させる。しかし、これは以下のセクションで繰り返し出てくる概念であるが、老化に対する防御反応は、最終的には有害となり、老化を悪化させる危険性がある。PI3KキナーゼやAKTキナーゼのマウスヌル変異が胚致死であることが例証するように(Renner and Carnero, 2009)、IISシグナル伝達レベルが極端に低いと、生命維持が困難になる。また、IGF-1レベルが非常に低いプロジェロイドマウスも存在し、このマウスではIGF-1を補充することで早期老化を改善することができる(Mariño et al.、2010)。
②その他の栄養感知システム:mTOR、AMPK、サーチュイン
高濃度のアミノ酸を感知するmTOR、高濃度のAMPを感知して低エネルギー状態を感知するAMPK、そして高濃度のNAD+を感知して低エネルギー状態を感知するサーチュインである(Houtkooper et al.)。
mTORキナーゼは、mTORC1とmTORC2という2つの多タンパク質複合体の一部であり、同化代謝の本質的なあらゆる側面を制御している(Laplante and Sabatini, 2012)。酵母、ミミズ、ハエにおいてmTORC1活性を遺伝的にダウンレギュレーションすると、長寿が延長し、DRによる更なる長寿効果が減弱することから、mTOR阻害はDRを模倣することが示唆される(Johnson et al.、2013)。マウスでは、ラパマイシンによる治療も寿命を延ばし、哺乳類の寿命を延ばす最も強力な化学的介入と考えられている(Harrisonら、2009)。mTORC1活性は低いがmTORC2は正常な遺伝子改変マウスは寿命が延び(Lammingら、2012年)、S6K1(mTORC1の主要基質)を欠損したマウスも長寿である(Selmanら、2009年)。したがって、mTORC1/S6K1のダウンレギュレーションは、mTORとの関連において哺乳類の長寿の重要なメディエーターであると考えられる。さらに、マウスの視床下部ニューロンでは加齢に伴いmTOR活性が上昇し、加齢性肥満の一因となっているが、これは視床下部へのラパマイシンの直接注入によって回復する(Yang et al.) これらの観察結果は、IIS経路が関与する観察結果とともに、IISまたはmTORC1経路を介してシグナル伝達される強い栄養活性と同化活性が、老化を促進する主要な要因であることを示している。TOR活性の阻害は、明らかに老化に有益な効果をもたらすが、創傷治癒障害、インスリン抵抗性、白内障、マウスにおける精巣変性など、望ましくない副作用もある(Wilkinson et al.) したがって、TOR阻害による有益な作用と有害な作用が、どの程度切り離せるのかを見極めるためには、そのメカニズムを理解することが重要であろう。
他の2つの栄養センサーであるAMPKとサーチュインは、IISとmTORとは逆の方向に作用する。つまり、栄養が豊富で同化作用がある代わりに、栄養不足で異化作用があることを知らせる。従って、これらのアップレギュレーションは、健康的な老化を促進する。AMPKの活性化は代謝に様々な影響を及ぼし、驚くべきことにmTORC1を遮断する(Alersら、2012年)。ミミズやマウスにメトホルミンを投与すると、AMPKの活性化が寿命延長を仲介する可能性があることを示す証拠がある(Anisimovら、2011;Mairら、2011;Onken and Driscoll、2010)。寿命調節におけるサーチュインの役割については前述した(「エピジェネティック変化」参照)。さらに、SIRT1はPPARγコアクチベーター1α(PGC-1α)を脱アセチル化し、活性化することができる(Rodgersら、2005)。PGC-1αは、ミトコンドリア新生、抗酸化防御の強化、脂肪酸酸化の改善など、複雑な代謝反応を指揮する(Fernandez-Marcos and Auwerx, 2011)。さらに、SIRT1とAMPKは正のフィードバックループを形成し、低エネルギー状態の両センサーを統合的な反応に結びつけることができる(Price et al.)。
【ミトコンドリアの機能不全】
細胞や生物が老化するにつれて、呼吸鎖の効率は低下する傾向にあり、その結果、電子の漏出が増加し、ATPの生成が減少する(Green et al.、2011)(図4B)。ミトコンドリア機能不全と老化の関係は長い間疑われてきたが、その詳細を解明することは老化研究の大きな課題として残されている。
①活性酸素
老化のミトコンドリアフリーラジカル理論では、老化に伴ってミトコンドリアの機能障害が進行すると、活性酸素の産生が増加し、その結果、ミトコンドリアの劣化がさらに進み、細胞全体がダメージを受けると提唱している(Harman, 1965)。老化における活性酸素の役割を支持するデータはいくつもあるが、ここでは、老化のミトコンドリアフリーラジカル説の激しい再評価を余儀なくされた、ここ5年間の進展に焦点を当てる(Hekimi et al.) 特に影響を与えたのは、酵母や線虫において活性酸素の増加が寿命を延ばすという予期せぬ観察であった(Doonan et al., 2008; Mesquita et al.) 同様に、ミトコンドリアの活性酸素と酸化的損傷を増加させるマウスの遺伝子操作は老化を促進せず(Van Remmenら、2003;Zhangら、2009)、抗酸化防御を増加させたマウスは寿命延長を示さない(Pérezら、2009)、 2009)、そして最後に、ミトコンドリア機能を損なうが活性酸素を増加させない遺伝子操作は老化を促進する(Edgarら, 2009; Hionaら, 2010; Kujothら, 2005; Trifunovicら, 2004; Vermulstら, 2008)。これらのデータやその他のデータは、老化における活性酸素の役割について再考する道を開いた(Ristow and Schmeisser, 2011)。実際、活性酸素の有害作用に関する研究とは別に、細胞内シグナル伝達の分野では、生理的シグナルやストレス状態に応答して増殖や生存を引き起こす活性酸素の役割に関する確かな証拠が蓄積されている(Sena and Chandel, 2012)。活性酸素を、AMPやNAD+と同様の概念で、ストレスによって誘発される生存シグナルとみなせば、この2つの証拠は調和する。この意味で、活性酸素の主な作用は、代償的恒常性反応の活性化である。年齢が進むにつれて、細胞ストレスとダメージが増加し、生存を維持しようとROSレベルも並行して上昇する。ある閾値を超えると、活性酸素レベルは本来の恒常性維持の目的を裏切り、加齢に伴うダメージを緩和するどころか、むしろ悪化させるようになる(Hekimi et al.) この新しい概念的枠組みは、加齢に対する活性酸素のプラス、マイナス、あるいは中立的な影響に関して、一見相反する証拠に対応できるかもしれない。
②ミトコンドリアの完全性と生合成
DNAポリメラーゼγを欠損したマウスを用いた研究(Edgarら、2009;Hionaら、2010)に見られるように、ミトコンドリアの機能不全は活性酸素とは無関係に老化の一因となる可能性がある(「ゲノムの不安定性」参照)。例えば、ミトコンドリアの欠損は、ストレスに応答してミトコンドリアが透過しやすくなることでアポトーシスシグナル伝達に影響を与え(Kroemer et al., 2007)、活性酸素を介した、あるいは透過を促進した炎症性サイトカインの活性化を促進することで炎症反応を引き起こす(Green et al.) また、ミトコンドリアの機能障害は、ミトコンドリア外膜と小胞体との界面に影響を与えることで、細胞内シグナル伝達や臓器間クロストークに直接影響を与える可能性がある(Raffaello and Rizzuto, 2011)。
加齢に伴うミトコンドリアの生体エネルギー効率の低下は、ミトコンドリアの生合成の低下など、複数のメカニズムに集約される可能性がある。例えば、テロメラーゼ欠損マウスにおけるテロメアの減少の結果、PGC-1αとPGC-1βがp53を介して抑制される(Sahin and DePinho, 2012)。このミトコンドリアの減少は野生型マウスの生理的老化の際にも起こり、テロメラーゼの活性化によって部分的に逆転させることができる(Bernardes de Jesus et al., 2012)。SIRT1は、転写コアクチベーターPGC-1α(Rodgersら、2005)とオートファジーによる損傷ミトコンドリアの除去(Leeら、2008)が関与するプロセスを通して、ミトコンドリアの生合成を調節する。主要なミトコンドリア脱アセチル化酵素であるSIRT3は(Lombardら、2007)、呼吸鎖、トリカルボン酸サイクル、ケト生成、脂肪酸β酸化の構成要素を含むエネルギー代謝に関与する多くの酵素を標的としている(Giralt and Villarroya、2012)。SIRT3はまた、主要なミトコンドリアの抗酸化酵素であるマンガンスーパーオキシドジスムターゼを脱アセチル化することによって、活性酸素の産生速度を直接制御する可能性もある(Qiu et al.) これらの結果を総合すると、テロメアとサーチュインがミトコンドリア機能を制御し、加齢に伴う疾患に対して保護的な役割を果たしている可能性がある。
生体エネルギー学的欠陥の原因となるその他のメカニズムとしては、mtDNAにおける突然変異や欠失の蓄積、ミトコンドリアタンパク質の酸化、呼吸鎖(超)複合体の高分子組織の不安定化、ミトコンドリア膜の脂質組成の変化などが挙げられる、 分裂と融合の不均衡によるミトコンドリアダイナミクスの変化、欠損したミトコンドリアをタンパク質分解の対象とするマクロオートファジーのオルガネラ特異的形態であるマイトファジーによる品質管理の欠陥などである(Wang and Klionsky, 2011)。生合成の低下とクリアランスの減少によるミトコンドリアの損傷増加とターンオーバーの低下が組み合わさることで、老化が進行する可能性がある(図4B)。
興味深いことに、持久力トレーニングと交互空腹は、ミトコンドリアの変性を回避する能力によって健康寿命を改善する可能性がある(Castello et al.) このような有益な効果は、少なくとも部分的には、持久力トレーニングと絶食の両方が強力な誘因となるオートファジーの誘導によって媒介されるのではないかと推測したくなる(Rubinsztein et al.) しかし、オートファジーの誘導は、健康的なライフスタイルが老化を遅らせる唯一のメカニズムではないだろう。というのも、正確なDRレジームによって、さらに長寿経路が活性化される可能性があるからである(Kenyon, 2010)。
③ミトホルミシス
加齢に伴うミトコンドリアの機能異常はホルミシスとも関連しており、この概念は最近、多くの加齢研究において注目されている(Calabrese et al.) この概念によれば、軽度な毒性治療は、引き金となった損傷の修復を上回る有益な代償反応を引き起こし、実際に、損傷を受ける前の状態と比較して、細胞のフィットネスを向上させる。したがって、重度のミトコンドリア機能不全は病原性であるが、軽度の呼吸不全は、おそらくホルモン反応によるものであろうが、寿命を延ばす可能性がある(Haigis and Yankner, 2010)。ホルモン反応は、線虫で示されたように、ミトコンドリアが欠損している同じ組織でも、また離れた組織でも、ミトコンドリア防御反応を引き起こす可能性がある(Durieux et al.) メトホルミンやレスベラトロールのような化合物は、AMPレベルの上昇とAMPKの活性化を特徴とする低エネルギー状態を誘導する穏やかなミトコンドリア毒であるという説得力のある証拠がある(Hawley et al.) 重要なことに、メトホルミンは、AMPKとマスター抗酸化制御因子NRF2を介する代償的ストレス応答の誘導を通じて、線虫の寿命を延ばす(Onken and Driscoll, 2010)。最近の研究では、メトホルミンはミミズの腸内細菌叢の葉酸とメチオニン代謝を阻害することにより、ミミズの老化を遅らせることも示されている(Cabreiro et al.) 哺乳類では、メトホルミンを幼少期から投与するとマウスの寿命が延びる(Anisimov et al.) レスベラトロールとサーチュイン活性化物質SRT1720の場合、PGC-1α依存的に代謝障害を防御し、ミトコンドリア呼吸を改善するという説得力のある証拠がある(Baurら、2006;Feigeら、2008;Lagougeら、2006;Minorら、2011)が、レスベラトロールは通常の食事条件下ではマウスの寿命を延長しない(Pearsonら、2008;Strongら、2013)。長寿におけるPGC-1αの役割をさらに裏付けるものとして、PGC-1αの過剰発現は、ミトコンドリア活性の改善と関連してショウジョウバエの寿命を延ばすのに十分であるという観察がある(Reraら、2011年)。最後に、ミトコンドリアの脱共役は、遺伝的に脱共役タンパク質UCP1を過剰発現させるか、化学的脱共役物質2-4-ジニトロフェノールを投与することによって、ハエやマウスの寿命を延ばすことができる(Caldeira da Silvaら、2008; Fridellら、2009; Gatesら、2007; Mookerjeeら、2010)。
【細胞老化】
細胞老化は、定型化された表現型の変化と結びついた、細胞周期の安定した停止として定義することができる(Campisi and d'Adda di Fagagna, 2007; Collado et al. この現象は、元来Hayflickによって、連続継代培養したヒト線維芽細胞で報告された(Hayflick and Moorhead, 1961)。今日では、Hayflickによって観察された老化はテロメアの短縮によって引き起こされることがわかっているが(Bodnarら、1998)、このテロメア過程とは無関係に老化を引き起こす他の老化関連刺激も存在する。最も注目すべきは、非テロメアDNA損傷とINK4/ARF遺伝子座の脱抑制であり、両者とも年代的老化に伴って進行するが、老化を誘導することも可能である(Collado et al., 2007)。老化組織における老化細胞の蓄積は、DNA損傷などの代用マーカーを用いて推測されることが多い。組織における老化を同定するために、老化関連β-ガラクトシダーゼ(SABG)を直接用いた研究もある(Dimriら、1995)。注目すべきは、肝臓におけるSABGとDNA損傷の詳細かつ並行的な定量化で、若齢マウスでは合計約8%、超高齢マウスでは約17%の老化細胞が検出され、同等の定量データが得られた(Wang et al.) 皮膚、肺、脾臓でも同様の結果が得られたが、心臓、骨格筋、腎臓では変化は見られなかった(Wang et al., 2009)。これらのデータから、細胞の老化は、老化した生物のすべての組織に一般化された性質ではないことが明らかである。老化した腫瘍細胞の場合、厳重な免疫監視にさらされ、貪食作用によって効率的に除去されるという十分な証拠がある(Hoenicke and Zender, 2012; Kang et al.) 考えられるのは、加齢に伴う老化細胞の蓄積は、老化 細胞の生成速度の増加や除去速度の低下を反映している可能 性である。

(A) 細胞老化。若い生物では、細胞の老化は損傷した細胞の増殖を防ぎ、癌から保護し、組織の恒常性に貢献します。古い生物では、広範囲にわたる損傷と老化細胞のクリアランス不足がそれらの蓄積をもたらし、これは老化に寄与する組織恒常性に多くの有害な影響を及ぼします。
(B) 幹細胞の枯渇。造血幹細胞(HSC)、間葉系幹細胞(MSC)、衛星細胞、および腸上皮幹細胞(IESC)の枯渇の結果が例示されています。
(C) 細胞間通信の変化。加齢に伴う細胞間コミュニケーションの変化の例。
老化とともに老化細胞の数が増えることから、老化は老化の一因であると広く考えられてきた。しかし、この見解は、おそらく老化の第一の目的であろう、傷ついた細胞の増殖を防ぎ、免疫系による死滅の引き金を引くことを過小評価している。したがって、老化は、傷ついた細胞や発癌の可能性のある細胞から組織を取り除くのに貢献する、有益な代償反応である可能性がある。しかし、この細胞チェックポイントには、老化細胞を除去し、前駆細胞を動員して細胞数を再増強する、効率的な細胞入れ替えシステムが必要である。老化した生物では、この入れ替わりシステムが非効率的になったり、前駆細胞の再生能力を使い果たしたりして、最終的に老化細胞が蓄積し、その結果、損傷が悪化して老化の一因となる可能性がある(図5A)。
近年、老化細胞は、特に炎症性サイトカインとマトリックスメタロプロテアーゼに富み、「老化に伴う分泌表現型」と呼ばれる、分泌物の劇的な変化を示すことが分かってきた(Kuilman et al.) この炎症性分泌物は老化に関与している可能性がある(「細胞間コミュニケーション」参照)。
①INK4a/ARF遺伝子座とp53
DNA損傷に加え、過剰な有糸分裂シグナル伝達も老化と最も強く関連するストレスである。最近の報告では、老化を誘導することができる発癌性あるいは分裂促進性の変化が50以上挙げられている(Gorgoulis and Halazonetis, 2010)。このような様々ながん原性障害に応答して老化を引き起こすメカニズムの数も増えてきたが、最初に報告されたp16INK4a/Rb経路とp19ARF/p53経路が、一般的に最も重要な経路であることに変わりはない(Serrano et al.) p16INK4aのレベル(より低い程度ではp19ARFも)が、マウスとヒトの両方で分析された本質的にすべての組織の年代と相関していることを考慮すると、これらの経路の老化との関連性はさらに顕著になる(Krishnamurthyら、2004;Resslerら、2006)。我々は、組織間、生物種間、そして若い組織と老齢組織との間で平均して1桁の変動幅で、発現が年代的加齢とこれほど強固に相関する遺伝子やタンパク質を他に知らない。p16INK4aとp19ARFはどちらも同じ遺伝子座、INK4a/ARF遺伝子座にコードされている。300以上のゲノムワイド関連研究(GWAS)の最近のメタアナリシスでは、INK4a/ARF遺伝子座が、数種類の心血管系疾患、糖尿病、緑内障、アルツハイマー病など、加齢に伴う病態に最も多く遺伝的に関連しているゲノム遺伝子座であることが同定された(Jeck et al.) これらの観察から、INK4a/ARF遺伝子座は、ヒトの老化と老化に伴う病態を制御する遺伝子として、最もよく立証されている。未解決の重要な問題は、疾患関連INK4a/ARF対立遺伝子が機能獲得型か機能喪失型かを決定することである。
細胞の老化誘導におけるp16INK4aとp53の重要な役割から、p16INK4a誘導性老化とp53誘導性老化が生理的老化に寄与しているという仮説が支持されている。この見解によれば、p16INK4aとp53の老化促進活性は、腫瘍抑制におけるその利点に比べれば、許容できる程度の犠牲であろう。これを支持するものとして、広範かつ持続的な損傷によって老化が早まった突然変異マウスは、劇的なレベルの老化を示し、そのプロジェロイド表現型はp16INK4aやp53の除去によって改善される。これはBRCA1欠損マウス(Cao et al., 2003)、HGPSモデルマウス(Varela et al., 2005)、BubR1の低型突然変異による染色体安定性欠損マウス(Baker et al., 2011)でも同様である。しかし、他の証拠はより複雑な像を示唆している。p16INK4a、p19ARF、またはp53癌抑制因子が軽度かつ全身的に増加したマウスは、予想される老化促進作用とは対照的に、癌発生率の低さでは説明できない長寿を示す(Matheuら、2007、Matheuら、2009)。また、p53を除去すると、いくつかのプロジェロイド変異マウスの表現型が悪化する(Begus-Nahrmannら、2009;Murgaら、2009;Ruzankinaら、2009)。ここでもまた、老化について上述したように、p53とINK4a/ARFの活性化は、損傷細胞の増殖と老化や癌への影響を回避することを目的とした有益な代償反応とみなすことができる。しかしながら、損傷が広範囲に及ぶと、組織の再生能力は枯渇または飽和し、このような極端な条件下では、p53とINK4a/ARFの応答は有害となり、老化を加速させる可能性がある。
【幹細胞の枯渇】
組織の再生能の低下は、老化の最も明白な特徴の一つである(図5B)。例えば、造血能は加齢とともに低下し、その結果、適応免疫細胞の産生が減少する-免疫老化と呼ばれるプロセス-、貧血や骨髄性悪性腫瘍の発生率が増加する(Shaw et al.) マウスの前脳(Molofsky et al., 2006)、骨(Gruber et al., 2006)、筋繊維(Conboy and Rando, 2012)など、基本的にすべての成体幹細胞コンパートメントで、幹細胞の同様の機能低下が見つかっている。高齢マウスを用いた研究では、造血幹細胞(HSC)の細胞周期活性が全体的に低下していることが明らかにされており、高齢の造血幹細胞は若い造血幹細胞よりも細胞分裂が少ない(Rossi et al.) これはDNA損傷の蓄積(Rossiら、2007年)やp16INK4aのような細胞周期阻害タンパク質の過剰発現(Janzenら、2006年)と相関している。実際、古いINK4a-/-造血幹細胞は、古い野生型造血幹細胞と比較して、より優れた生着能を示し、細胞周期活性が増加している(Janzenら、2006)。テロメアの短縮もまた、様々な組織における加齢に伴う幹細胞の減少の重要な原因である(Floresら、2005;Sharpless and DePinho、2007)。これらは、幹細胞の衰退が複数の種類のダメージの統合的な結 果として現れるという、より大きなイメージの一例に過ぎない。
幹細胞や前駆細胞の不十分な増殖は、生体の長期的な維持にとって有害であることは 明らかであるが、幹細胞や前駆細胞の過剰な増殖もまた、幹細胞ニッチの疲弊を促進す ることによって有害となりうる。幹細胞の長期的な機能性にとって幹細胞の静止が重要であることは、ショウジョウバエの腸管幹細胞のケースで説得力のある形で証明されており、過剰な増殖は幹細胞の疲弊と早期老化を引き起こす(Rera et al.) 同様の状況はp21欠損マウスでも見られ、造血幹細胞や神経幹細胞が早期に疲弊してしまう(Cheng et al.、2000;Kippin et al.、2005)。この点で、加齢に伴うINK4aの誘導(「細胞の老化」参照)と血清IGF-1の減少(「調節された栄養感知」参照)は、どちらも幹細胞の静止状態を維持しようとする生体の試みを反映しているのかもしれない。また、最近の研究では、老化した筋幹細胞ニッチでFGF2シグナルが増加すると、静止状態が失われ、最終的には幹細胞の枯渇と再生能力の低下が起こるが、このシグナル伝達経路を抑制すると、これらの欠損が救済されることが示されている(Chakkalakal et al.) このことは、加齢による幹細胞の枯渇を抑えるために、FGF2シグナル伝達を阻害することを目的とした戦略をデザインする可能性を開く。この線に沿って、DRは腸や筋肉の幹機能を高めることが報告されている(Cerlettiら、2012;Yilmazら、2012)。
幹細胞の機能低下に関する重要な議論は、細胞内在性の経路と細胞 外在性の経路の相対的な役割である(Conboy and Rando, 2012)。最近の研究は、後者を強く支持している。特に、若いマウスの筋肉由来幹細胞をプロジェロイドマウスに移植すると、寿命が延び、ドナー細胞が検出されない組織においても、プロジェロイドマウスの退行性変化が改善されることから、その治療効果は分泌因子による全身的効果に由来する可能性が示唆されている(Lavasani et al.) さらに、パラビオ ーシス実験により、老齢マウスにおける神経幹細胞や筋幹細胞 の機能低下は、若齢マウスからの全身性因子によって回復することが明ら かになっている(Conboyら、2005;Villedaら、2011)。
幹細胞機能を改善するための薬理学的介入も研究されている。特に、ラパマイシンによるmTORC1阻害は、プロテオスタシスを改善し(「プロテオスタシスの喪失」参照)、エネルギー感知に影響を与えることで(「調節された栄養感知」参照)、老化を先延ばしすることができ、表皮、造血系、腸における幹細胞機能も改善する可能性がある(Castilhoら、2009;Chenら、2009;Yilmazら、2012)。このことはまた、ラパマイシンの抗老化活性の機序的根拠を明確にすることの難しさを示しており、ここで論じた老化のさまざまな特徴の間に相互に関連性があることを強調している。また、老化した造血幹細胞で活性が亢進しているGTPase CDC42を薬理学的に阻害することで、ヒトの老化細胞を若返らせることが可能であることも特筆に値する(Florian et al.)。
【細胞間コミュニケーションの変化】
細胞自律的な変化だけでなく、老化は、内分泌、神経内分泌、神経細胞など、細胞間情報伝達レベルでの変化も含んでいる(Laplante and Sabatini, 2012; Rando and Chang, 2012; Russell and Kahn, 2007; Zhang et al. このように、神経ホルモンシグナル伝達(例えば、レニン-アンジオテンシン、アドレナリン作動性、インスリン-IGF1シグナル伝達)は、炎症反応が増加し、病原体や前悪性腫瘍細胞に対する免疫監視機能が低下し、細胞周囲および細胞外環境の構成が変化するにつれて、加齢に伴い調節されなくなる傾向がある。
①炎症
加齢に伴う細胞間情報伝達の顕著な変化は、「炎症老化」、すなわち哺乳類において加齢に伴って生じる、くすぶり続ける炎症性表現型である(Salminen et al., 2012)。炎症は、炎症性組織損傷の蓄積、機能不全に陥った免疫系が病原体や機能不全に陥った宿主細胞を効果的に排除できないこと、老化細胞が炎症性サイトカインを分泌しやすくなること(「細胞老化」参照)、NF-κB転写因子の活性化が亢進すること、あるいはオートファジー反応に欠陥が生じることなど、複数の原因によって生じる可能性がある(Salminen et al.) これらの変化は、NLRP3インフラマソームや他の炎症性経路の活性化を高め、最終的にIL-1β、腫瘍壊死因子、インターフェロンの産生を増加させる(Greenら、2011;Salminenら、2012)。炎症はまた、肥満と2型糖尿病の発症にも関与しており、この2つの病態はヒト集団における老化の一因であり、また老化と相関している(Barzilai et al., 2012)。同様に、炎症反応の欠陥はアテローム性動脈硬化症に重要な役割を果たしている(Tabas, 2010)。加齢に伴う炎症が表皮幹細胞の機能を阻害するという最近の発見(Doles et al. 炎症老化と並行して、適応免疫系の機能も低下する(Deeks, 2011)。この免疫老化は、免疫系が感染物質や感染細胞、悪性化寸前の細胞を排除できないために、全身レベルで老化の表現型を悪化させる可能性がある。さらに、免疫系の機能の一つは、老化組織や前悪性病変に蓄積する多倍体細胞と同様に、老化細胞(「幹細胞の枯渇」を参照)を認識して排除することである(Davoli and de Lange, 2011; Senovilla et al.)。
加齢組織の転写様式に関する世界的な研究でも、加齢における炎症経路の重要性が強調されている(de Magalhães et al.) NF-κB経路の過剰活性化は、老化の転写シグネチャーの一つであり、トランスジェニックマウスの老化した皮膚にNF-κB阻害剤を条件付きで発現させると、この組織の表現型が若返り、若い年齢に対応する転写シグネチャーが回復する(Adlerら、2007年)。同様に、NF-κBシグナル伝達を遺伝的・薬理学的に阻害することで、様々な老化促進モデルマウスにおいて加齢に伴う特徴が抑制される(Osorio et al.) 炎症と老化の新たな関連は、炎症やストレス応答が視床下部においてNF-κBを活性化し、ニューロンによる性腺刺激ホルモン放出ホルモン(GnRH)の産生低下をもたらすシグナル伝達経路を誘導するという最近の発見に由来する(Zhang et al.) このGnRHの減少は、骨の脆弱性、筋力低下、皮膚の萎縮、神経新生の低下など、多くの老化に関連した変化の原因となる。一貫して、GnRH治療は老化による神経新生の障害を防ぎ、マウスにおける老化の進行を減速させる(Zhangら、2013年)。これらの所見は、視床下部がNF-kBを介した炎症反応とGnRHを介した神経内分泌作用を統合することによって、全身の老化を調節している可能性を示唆している。
炎症と老化を関連付けるin vivoでのさらなる証拠は、サイトカインのmRNA分解を仲介することで炎症反応の停止に関与するmRNA崩壊因子AUF1に関する研究から得られた(Pont et al.) AUF1欠損マウスは顕著な細胞老化と早期老化の表現型を示すが、このRNA結合因子を再発現させることで回復させることができる。興味深いことに、AUF1は炎症性サイトカインのmRNA分解を誘導することに加えて、テロメラーゼ触媒サブユニットTERTの発現を活性化することによってテロメアの長さの維持にも寄与しており(Pont et al. )
加齢に伴う炎症反応にも影響を及ぼしている可能性がある。いくつかの研究から、ヒストンとNF-κBなどの炎症シグナル伝達経路の構成要素を脱アセチル化することにより、SIRT1は炎症関連遺伝子をダウンレギュレートすることが明らかになっている(Xie et al.) これらの知見と一致して、SIRT1レベルの低下は多くの炎症性疾患の発症と進行と相関しており、SIRT1の薬理学的活性化はマウスにおける炎症反応を予防する可能性がある(Gillumら、2011;Yaoら、2012;Zhangら、2010)。SIRT2とSIRT6もまた、NF-kBサブユニットの脱アセチル化と標的遺伝子の転写抑制を通じて、炎症反応を抑制する可能性がある(Kawahara et al., 2009; Rothgiesser et al., 2010)。
②その他の細胞間コミュニケーション
炎症以外にも、ある組織における加齢に関連した変化が、他の組織の加齢に特異的な劣化につながることを示す証拠が蓄積しており、加齢表現型の臓器間連携を説明している。炎症性サイトカイン以外にも、「伝染性老化」やバイスタンダー効果の例があり、老化細胞がギャップジャンクションを介した細胞間接触や活性酸素を介したプロセスを通じて、隣接する細胞の老化を誘導する(Nelson et al.) マウスの養子移入モデルを用いて評価したように、微小環境はCD4 T細胞の加齢に伴う機能障害に寄与している(Lefebvre et al.) 逆に、ある一つの組織を標的とした寿命延長操作は、他の組織の老化プロセスを遅らせることができる(Durieuxら、2011;Lavasaniら、2012;Tomás-Lobaら、2008)。
③欠損した細胞間コミュニケーションの回復
加齢に伴って失われる細胞間情報伝達特性を改善するような遺伝的、栄養学的、薬理学的介入など、加齢過程の根底にある欠陥のある細胞間情報伝達を回復させる可能性はいくつかある(Freije and López-Otín, 2012; Rando and Chang, 2012)。この点で特に興味深いのは、健康寿命を延ばすためのDRアプローチ(Piperら、2011;Sanchez-Romanら、2012)と、パラビオシス実験で同定された血液由来全身性因子の使用に基づく若返り戦略(Conboyら、2005;Loffredoら、2013;Villedaら、2011)である。さらに、アスピリンなどの抗炎症剤を長期投与することで、マウスでは寿命が延び、ヒトでは健康的な老化が促進される可能性がある(Rothwellら、2011;Strongら、2008)。さらに、腸内細菌叢が宿主の免疫系の機能を形成し、全身的な代謝効果を発揮していることを考えると、人体の複雑でダイナミックな腸内細菌生態系の組成と機能性を操作することによって、寿命を延ばすことが可能であると考えられる(Claesson et al.)。
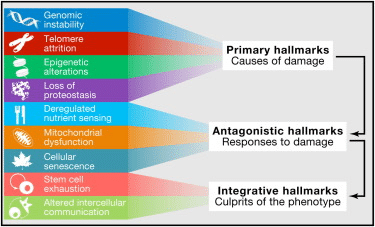
提案された9つの老化の特徴は、3つのカテゴリに分類されます。(上)これらの特徴は、細胞損傷の主な原因と考えられています。(中間)損害に対する補償的または拮抗的な反応の一部と見なされるもの。これらの反応は最初は損傷を軽減しますが、最終的には慢性的または悪化した場合、それ自体が有害になります。(下)統合的特徴は、前の2つの特徴グループの最終結果であり、老化に関連する機能低下の最終的な原因となります。

老化の9つの特徴は、マウスの原則の証拠があるそれらの治療戦略とともに示されています。