
Rを使ったクラスタリング樹形図の作成手順と注意事項② Rを動かしてクラスタリングする

前回まででデータの下準備が完了しました。Rで読み取れるようにするためCSVファイルに変更したり、R1C1形式を採用したり...
今回は加工したデータをRのコマンドを使ってクラスタリングします。この記事ではクラスタリングの理論については触れず、ただクラスタリング結果を出力するための流れを説明します。理論は別の記事で説明しようと思うのでそちらもご覧ください。それでは始めましょう。
1.ディレクトリの指定
Rでは参照するデータファイルをどこから引っ張ってくるかを指定する必要があります。フォルダ1に参照したいデータが入っているのにフォルダ2を指定しているといくら頑張っても呼び出せません。
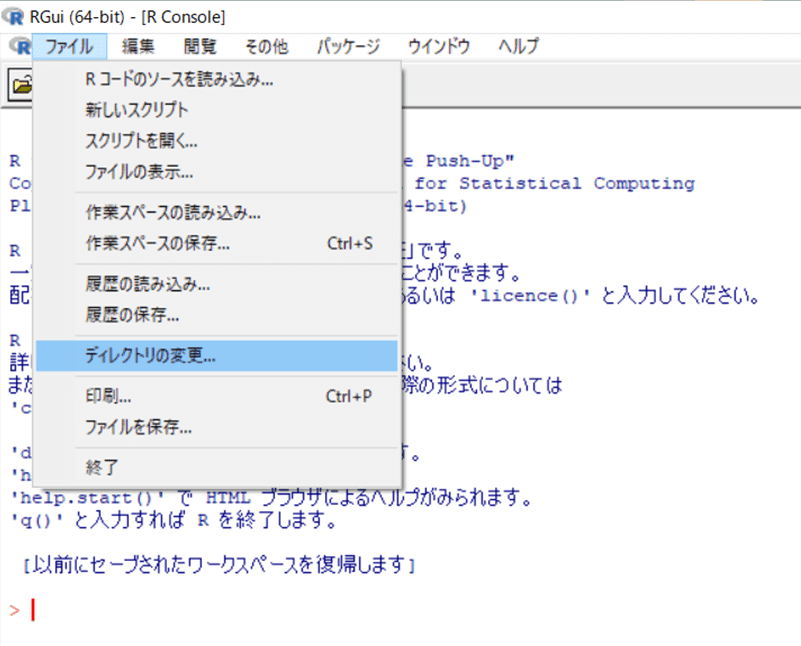
まずRを立ち上げたら「ファイル」→「ディレクトリの変更」をクリックします。
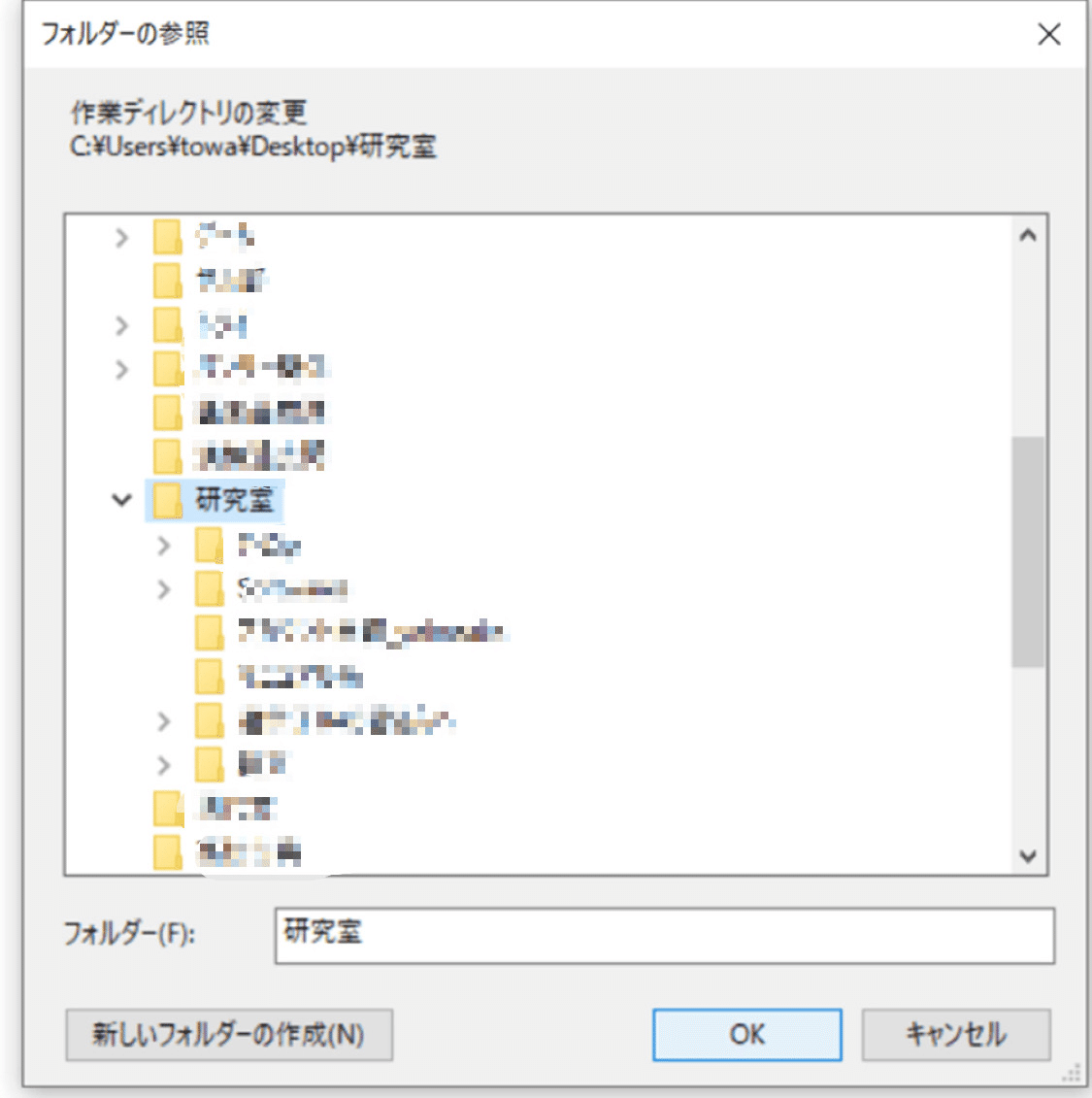
そうするとこのような画面が出るので、データが入っているフォルダを指定します。僕の場合は「研究室」フォルダにデータを突っ込んでいるのでそれを指定します。もちろんデスクトップにおいてある場合はデスクトップを指定することもできます。
2.必要なパッケージのインストール
Rは統計解析に特化したプログラムですが、クラスタリング樹形図やヒートマップといったものを作成するためのプログラムは最初から入っているわけではありません。なので使いたいプログラムをあらかじめインストールしておくことが必要になります。Rで使えるような種々のプログラムのことを"パッケージ"と呼びます。(もちろんあらかじめ入っているものもあります)
必要なパッケージをインストールする場合以下のように打ち込みます。これは全てのパッケージで共通なので必ず覚えましょう。
install.packages("パッケージ名")これを入力すると国名が羅列されたタブが表示されますが、これは「どこの国経由でインストールするか」を選ぶものになります。僕は日本にいるので「Japan」を選択してます。自分がいる場所に最も近い所を選べばOKです。
今回必要なパッケージは以下の通りで、これをそのままRに張りつければすべて同時にインストールできます。
install.packages("dplyr")
install.packages("ggplot2")
install.packages("cluster")
install.packages("fastcluster")
install.packages("microbenchmark")
install.packages("gplots")
install.packages("dendextend") 今回はこのうちの一部を使って樹形図を作成するのですべて必要というわけではありませんが、その他の手法を使ったクラスタリングを行うときに使用するのでまとめてインストールしておくことをお勧めします。
また、ものによってはインストールが上手くいかない場合もあるので、そのときのトラブルシューティングとして以下のサイトを参照してください。
3.コマンドを打ち込んでクラスタリング結果を表示する
以下のコマンドを打ち込めば表示できます。具体的な理屈は別記事で書くとして、大まかにコマンドの意味を説明します。
> x <- read.csv("sample1.csv", header=T, row.names=1)
> x2 <- as.matrix(x)
> x3 <- cor(x2, method="spearman")
> d <- as.dist(1-x3)
> h <- hclust(d, method="ward.D2")
> plot(h) まず1行目でデータのファイルを読み込んでいます。「read.csv」でcsvファイルを読み取らせようとしていて、「"sample1.csv"」で読み込むファイルを指定しました。
「header=T」ですが、これは「データの名前が一行目に書いてあるから数値としては読み込まないでね」というコマンドです。データを見ると1行目はtissue1~5が書いてあるのでこのコマンドを入れています。もしデータ名の記載がないようなものであれば「header=F」と入力します。このTとFはもちろんTRUEとFALSEの頭文字です。
「row.names=1」も似たようなもので、「1列目を列名として指定する」というものです。データが2列目から始まっていれば「row.names=2」と入れればOKです。今回は予めデータを左上に寄せていたので、1を入力しています。
最後に一番左の「x<- 」についてです。これは「xという文字にデータを格納する」という意味です。これより右のコマンドで読み込んだファイルがあって、それにxという名前を付けて保存するイメージです。xという文字の中にsample1のデータが格納されているわけです。「<-」は多分矢印のイメージだと思います。今回は「x」という文字に格納しましたが、別に格納先の文字は何でもいいので、他のRを解説しているところでは「data<-」とか「dx<-」とか書いてあったりします。どれもやってることは全く一緒です。
2行目以降の「x2<-」「d<-」も同じことをやっていて、その中に加工していったデータを一端保存しています。当然文字を変えてもOKです。
2行目の「x2 <- as.matrix(x)」は「xのデータを行列として扱ってx2にしまえ」というコマンドです。xには先ほどのデータが入っていますが、実際に演算を行うにはそれを行列として扱う必要があります。そのためのコマンドが2行目でやっていることです。
3行目は「x3 <- cor(x2, method="spearman")」です。ここでは「類似度」の計算を行っています。クラスタリングは「似ている」もの同士を同じグループにまとめていくものですから、それぞれのデータの類似度を計算する必要があるわけです。ここではx2に入っているデータをスピアマンの順位相関係数を用いて類似度を出しています。「method="spearman"」の中身を変更すれば別の類似度を用いることもできます。別の類似度については理屈を説明する記事で書こうかなぁと思ってます。
4行目は計算したスピアマンの順位相関係数を用いて距離を算出しています。スピアマンの順位相関係数は二つのデータが似ていれば1に近づき、似ていなければ-1に近づきます。なので「1-相関係数」を計算したとき、似ているほどその値が小さくなるようになっています。これを距離として扱うのが「as.dist」の部分です。絶対値が小さいものを近い=似ているとしています。
そして5行目でクラスタリングを行います。「hclust」というパッケージを使用しています。注目してほしいのが「method="ward.D2"」の部分で、ここでクラスタリングに用いる手法を指定しています。ウォード法は広く一般に用いられるクラスタリングの手法なので覚えておくとよいでしょう。もちろん、ここを書きかえれば別の手法でクラスタリングできます。
そして6行目「plot(h)」でクラスタリングの樹形図を表示することができます。以下にそれを示します

結果はこうなります。tissue2とtissue4は非常によく似ていて、tissue3は他のどれとも似ていないことを意味しています。このようなステップでクラスタリングができます。
4.注意事項
今回はスピアマンの順位相関係数やウォード法を使用しましたが、あくまでこれらは手法の一つであって、実験の目的や条件によっては不適切である場合もでてきます。したがって、他の手法とどのような違いがあるのかをしっかり理解しておくことがこういったバイオインフォマティクスの操作を行うときに重要になってくるわけです。具体的な各手法については別の記事で解説したいなと考えています。
5.遺伝子ごとにクラスタリングしたい場合
今回の例では「組織」をクラスタリングしましたが、「遺伝子」をクラスタリングしたい場合はどうすればよいのでしょう。結論を言うと、行列を転置するだけです。転置とは行と列を入れ替える操作なので、これさえやればあとは同じです。転置するコマンドは
> data <- t(matrix)転置したい行列データの前に「t」を付けるだけです。なんて簡単なんだろう。結果を以下に示します。
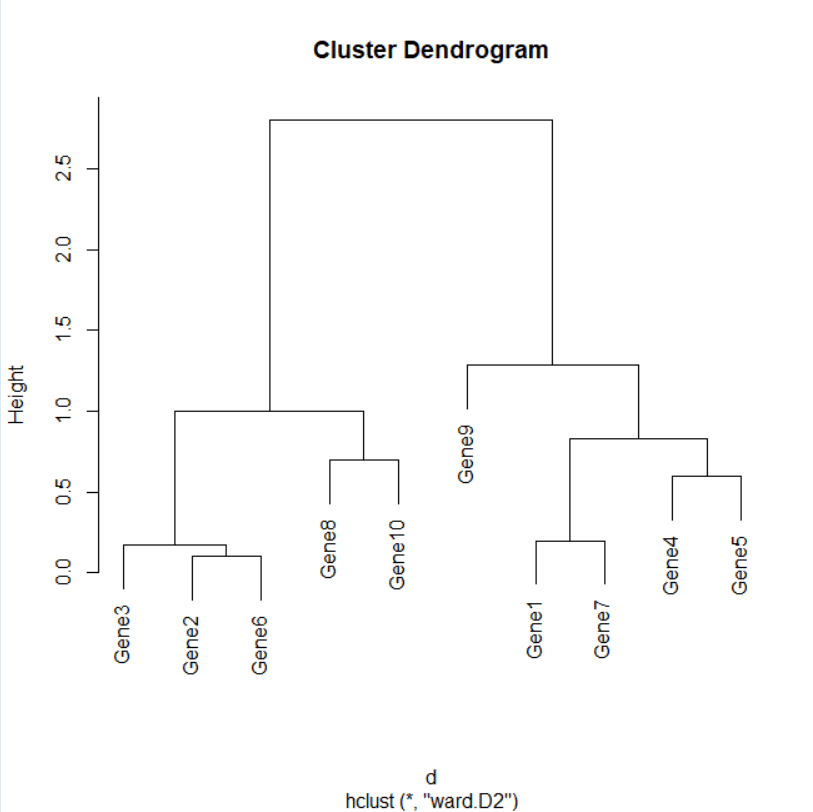
以上でR上でのクラスタリング樹形図の作成チュートリアルを終えます。次回は数理モデルについて注意事項も含めて書いていきます。
この記事が気に入ったらサポートをしてみませんか?