
【Aidemy X Bio】1222例の乳がんデータを深層学習させてみる!!(1):データ可視化編

前に
【Aidemy X Bio】がん細胞データをつかって、機械学習最先端手法Constrastive PCAをつかってみる
というお題でがん細胞の遺伝子発現データを用いて、一連の機械学習の分析を行わせてきた。その時はトータル20例の遺伝子発現データをつかっていわばお遊び的に分析を行ってきた。
今回試みようとしているのは、アメリカが国家プロジェクトとして行っているTCGAというがんのデータベースから遺伝子研究のサンプルデータを引っ張ってきて解析しようというもの。
これはRNAseqといって細胞内のRNAの配列を読み、正常、がんの各種細胞集団の中に、どの遺伝子がどのくらい使われているかを検出したものであり、今回情報の多い乳がんのデータに注目して行おうと思っているが、サンプル数が数千と途端に規模が大きくなりハードルも高くなる!
TCGAデータベースからのデータダウンロード

対象の乳がんを選択

ファイルのリポジトリをみて

transcriptome profiling, RNAseqを選ぶ
嬉しいことに生データでなく、カウントファイルをtxtデータで落とせるのがここのサイトのよいところだ。
正規化手法には議論はあるため、本当はcountファイルを落とすのが理想的なのであるが、FPKMという正規化されたカウントデータを落とすことにする

この時データーだけでなく、metadata fileとclinical dataファイルを落としておくといい。
データファイルはgdc_download_20180802_032509.tar.gzというような圧縮されたフォルダにサンプルごとのフォルダに圧縮されて入っており、0c7d119e-22c0-47ed-b736-3404ab2c7065.FPKM.txt.gzのような名前がついていて、1092症例、1222サンプル(一部同一患者の複数のサンプルが含まれている)からなる膨大なものであり。各々60483遺伝子の情報が含まれている。
正常の部位(Solid Normal Tissue)、がんの原発部位(Primary Tumor)、がんの転移部位(Metastatic )の3種のほか、患者さんの状況が、初期のがん(Stagei)から末期のがん(Stageiv)まで含まれているために、深層学習するには面白いはずだ!!
1222のサンプルがあるためデータファイルの解凍はunixを用いて行ったので詳細は割愛する。
そのあとRを用い1222のサンプルのデータをひとつのスプレッドシートに結合した(後で詳細書きます)
とまあここまでは順調に進んでいるように見えるが、ローカルマシーンで1220X60483の行列を作る際にメモリー不足に陥りあえなく断念!しかたないので4分割してファイルを作成した。
60483の遺伝子のうち検出できていないものも多くそのような遺伝子を多く含んでいると特徴が隠れているのでフィルターして除くことにする。
具体的にはファイル1で各サンプルの値が1以上であるものが150サンプル(約半分)以上ある遺伝子にのみ注目することにしてフィルターをかけて除去する。これで13231遺伝子に絞れる。その処理を他の4ファイルにも行う。そのあとでFPKMはlog2処理(log2(FPKM+1))をすることで正規分布二近くなり扱いやすいようなので、その処理を行って、ファイルをアウトプットしておわり。
#ファイル1処理
x <- read.table("b1.txt", sep="\t", header=T, row.names=1)
dim(x)
head(x)
head(x[,1])
x <- data.frame(x[])
#データーのフィルタリング
keep <- rowSums(x[, 2:ncol(x)]>1) > 150
keep
y <- x[keep,]
dim(y)
a <- y[,2:ncol(y)]
a <- log(a+1)
rownames(a) <- y[,1]
dim(a)
write.table(a, file="b1_mod.txt", sep="\t")
#ファイル2処理
x2 <- read.table("b2.txt", sep="\t", header=T, row.names=1)
head(x2)
dim(x2)
y2 <- x2[keep,]
dim(y2)
a2 <- y2[,1:ncol(y2)]
a2 <- log(a2+1)
rownames(a2) <- y[,1]
dim(a2)
write.table(a2, file="b2_mod.txt", sep="\t")
#ファイル3処理
x3 <- read.table("b3.txt", sep="\t", header=T, row.names=1)
head(x3)
dim(x3)
y3 <- x3[keep,]
dim(y3)
a3 <- y3[,1:ncol(y3)]
a3 <- log(a3+1)
rownames(a3) <- y[,1]
dim(a3)
write.table(a3, file="b3_mod.txt", sep="\t")
#ファイル4処理
x4 <- read.table("b4.txt", sep="\t", header=T, row.names=1)
head(x4)
dim(x4)
y4 <- x4[keep,]
dim(y4)
a4 <- y4[,1:ncol(y4)]
a4 <- log(a4+1)
rownames(a4) <- y[,1]
dim(a4)
write.table(a4, file="b4_mod.txt", sep="\t")いよいよこれらのファイルをつかって解析するわけであるが、ローカルマシーンでメモリ不足が起こってしまうので、googlecolaboratoryのGPUマシンを利用することとした!
Google Colaboratory
Google ColaboratoryとはGoogleの研究活動のひとつで、GPUマシンをgoogleアカウントのある人ならば誰でも無料で使える試みである。
環境構築なしでjupyter notebookでpythonがつかえるので、深層学習を勉強している人にはうってつけだ!
必要なライブラリを読み込んで、ファイルを読み込んで
%matplotlib inline
from sklearn.cluster import AffinityPropagation, KMeans, DBSCAN, SpectralClustering
from sklearn.manifold import MDS, TSNE, Isomap
from sklearn.metrics import silhouette_score
import numpy as np
import matplotlib.pyplot as plt
import pandas as pd
from scipy.linalg import logm, expmfrom google.colab import files
f=files.upload()
準備完了!
ファイルを読み込んで、形がOKかチェック
b1 = pd.read_csv("b1_mod.txt", sep="\t", header=0, index_col=0)
b1.shape
(13231, 299)これを4つのファイル全てに行い、ファイルを結合
df_wine = pd.concat([b1,b2,b3,b4],axis=1)
df_wine
形がOKなのでtxtファイルに落としておく。
扱いやすいようにデーターを転置し、必要なデータをXに格納
df_wine_2 = df_wine.T
#print(df_wine_2)
df_wine_2.shape
X = df_wine_2.iloc[0:, 1:].valuesメタファイル上でどのサンプルが、がんでどれが正常化わかっているので、そのラベルをあらかじめ作成しておいて読み込む
c = pd.read_csv("type.txt", sep="\t", header=0)
c = c.T
d = c.iloc[0,:].valuesまずPCAに近いというIsomapで可視化
ISO = Isomap()
ISO_coords = ISO.fit_transform(X)
plt.scatter(ISO_coords[:,0], ISO_coords[:,1])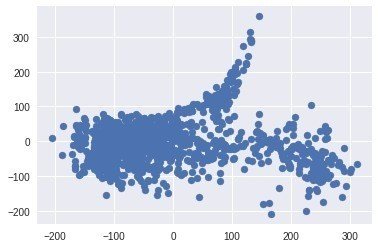
なんか思わせぶりな形!
正常(赤)とがん(オレンジが原発巣、緑が転移)のラベルを合わせてみると
pre_cluster_lables =d
print(len(pre_cluster_lables))
#pre_cluster_lables
cmap = plt.cm.get_cmap('prism', len(pre_cluster_lables))
cmap
plt.scatter(ISO_coords[:,0], ISO_coords[:,1], c=pre_cluster_lables, cmap=cmap)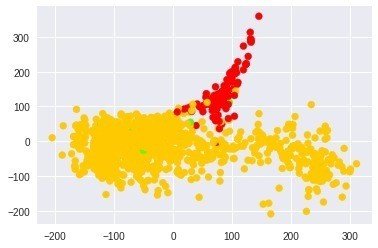
あれ、結構うまく分かれていそう!!
思わずクラスタリングさせてみると
km = KMeans(n_clusters=6)
km_cluster_labels = km.fit_predict(X)
km_cluster_labels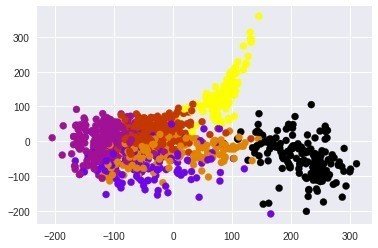
面白い感じに分かれている!
同様にTSNEプロットも試してみる
TS = TSNE()
TS_coords = TS.fit_transform(X)
plt.scatter(TS_coords[:,0], TS_coords[:,1])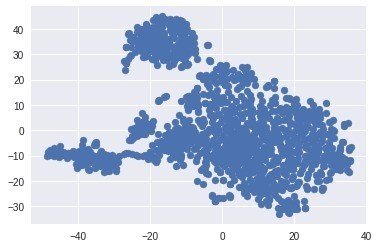
なかなか面白いパターン
上と同様に正常かがんで色分け
pre_cluster_lables =d
print(len(pre_cluster_lables))
#pre_cluster_lables
cmap = plt.cm.get_cmap('prism', len(pre_cluster_lables))
plt.scatter(TS_coords[:,0], TS_coords[:,1], c=pre_cluster_lables, cmap=cmap)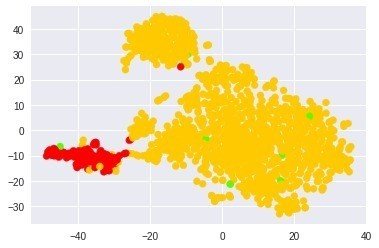
やはり面白いパターン
クラスタリングしてみても
cmap = plt.cm.get_cmap('gnuplot', len(km_cluster_labels))
plt.scatter(TS_coords[:,0], TS_coords[:,1], c=km_cluster_labels, cmap=cmap)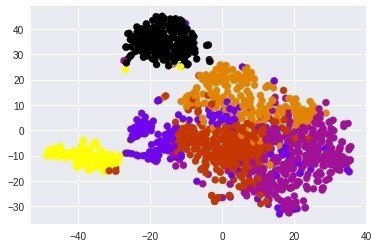
ちょっと面白いパターンですね。幾つかのクラスターが分かれていそう。
おそらくがんと正常、それからがんの中のサブクラスがうまくわかれているのではないだろうか?メタデータファイルやクリニカルデータファイルにさらに情報が乗っているのでそれらと突き合わせるともっと興味深いことがわかるかもしれない。またがんと正常、がんの中の初期と末期なども深層学習させて予測することも可能かもしれない!楽しみなデータである。
うまくいったとこだけのせてますが、実はスモールスケールでいろいろ試してました。この時はうまくいっていなかったな。。笑
なお最後に補足であるが、FPKMという正規化はPCAやtSNEなどの手法にいまひとつ適していないという意見もある(1)。一方でlog2(FPKM+1)などの値が論文などで散見されるので、 使用してもあながち間違いではないと思われる(2)が、綺麗にPCA, tSNEをおこなうためにはTMM(3)やRLE(4)といった正規化手法をためす必要があるかもしれない。
*1 https://www.biostars.org/p/219883/
https://www.nature.com/articles/srep23270/figures/2
https://www.nature.com/articles/srep28400/figures/1
*3 https://bi.biopapyrus.jp/rnaseq/analysis/normalizaiton/tmm.html
http://kazumaxneo.hatenablog.com/entry/2017/07/17/125137
*4 https://www.bioconductor.org/help/course-materials/2016/CSAMA/lect-06-rnaseq-analysis/rnaseq.pdf
*5 https://www.biostars.org/p/272304/