STARでRNA-seqデータのマッピング1

まず、インデックスファイルを作ります。
この処理はメモリをたくさん使うので、哺乳類は16GB以上、推奨は32GB以上だそうです。
時間がかかることを覚悟して実行してみます。
(実行するPCは、16GB)
まず、インデックスファイルを出力するSTAR_referenceディレクトリをつくります。
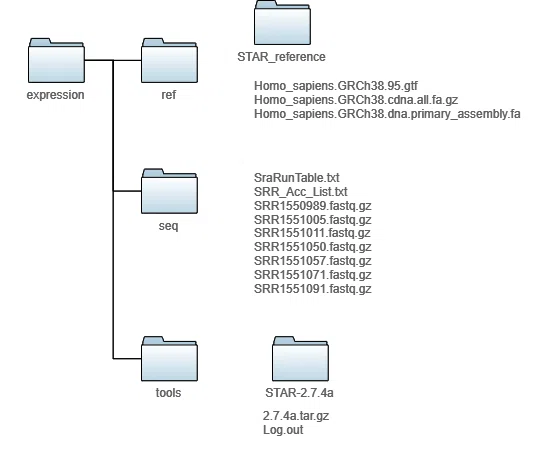
いよいよ、インデックスファイルを作ります。
コマンド STAR --runMode genomeGenerate --genomeDir ..ref/STAR_reference --genomeFastaFiles ../ref/Homo_sapiens.GRCh38.dna.primary_assembly.fa --sjdbGTFfile ../ref/Homo_sapiens.GRCh38.95.gtf
--runMode genomeGenerate インデックスの作成
--genomeDir インデックスファイルを入れるディレクトリ
--genomeFastaFiles リファレンスゲノムのFastaファイルの指定(Path)
--sjdbGTFfile アノテーションファイル
マニュアルhttps://github.com/alexdobin/STAR/blob/master/doc/STARmanual.pdf

実行しましたが、16時50分にランして1時になっても終わらなかったので
強制的に落としました。あまりにも長すぎるので調べてみると、
–runThreadNを指定すると、マルチスレッドで実行。使用するスレッド数を指定することができるそうなので、とりあえず10を指定してみました。
コマンド STAR –runThreadN 10 --runMode genomeGenerate --genomeDir ..ref/STAR_reference --genomeFastaFiles ../ref/Homo_sapiens.GRCh38.dna.primary_assembly.fa --sjdbGTFfile ../ref/Homo_sapiens.GRCh38.95.gtf

16時8分にランして18時43分に無事終了したした。
約2時間半。
タスクマネージャーでメモリーの使用量やCPUの使用量を見ていたのですが
ほぼ100%になっていたので、16GBだとスレッド数は10ぐらいかなと思います。
メモリを64GBぐらいのせるて実行してみたいです。
つぎは、いよいよマッピングです!