Rでheatmapを作成

マイクロアレイ解析やオミックス解析でよく見かけるheatmap。
下記サイトを参考にheatmapの描き方を勉強したのでメモ。
だれでもできるマイクロアレイ解析
http://blogs.yahoo.co.jp/doshiroutomicroarray
まず、Rを起動します。
Bioconductorがインストールされていない場合には、インストール。
(2024年6月時点でbioconductorのインストール方法が変わっていたので、文末に追記しました。https://bioconductor.orgと追記を参照のこと。)
> source("http://bioconductor.org/biocLite.R")
> biocLite()genefilterやgplotsなどのパッケージがインストールされていない場合は、以下のコマンドでRにインストール
> source("http://bioconductor.org/biocLite.R")
> biocLite("genefilter")> source("http://bioconductor.org/biocLite.R")
> biocLite("gplots")まず、Rを起動して、作業ディレクトリをデータのあるフォルダへ変更。
おまじない。
> library(genefilter)
> library(gplots)gtoolsやgdataがダウンロードされるので待ちます。
今回は、デモとして以下のファイルを使ってみましょう。
このファイルは、シロイヌナズナ時計遺伝子群の系時的な発現変動を示したものです。
clockdemo.txtの中身は、以下のテキストをコピペして作成することもできます(スペースはTabにして、Tab区切りテキストとして保存)。
26 30 34 38 42 46 50 54 58 62 66 70 74
TOC1/PRR1 541.9 669.4 2826.5 2302.9 1496.5 873.4 684.3 1307.4 2638.3 2284.9 1698.8 982 813.4
PRR3 348.7 409.9 1213.3 1460.3 843.9 457.9 407.4 684.6 1549.8 1077.3 784 722.3 559.6
PRR5 154.1 554 1269.4 539.5 427.8 250.3 178.6 1004.2 995.6 568.8 548.3 286.3 291.4
PRR7 436.1 1254.8 1307.6 220.4 25.5 120.6 681.4 1641 911.5 188.1 113.5 288.4 634.3
LHY 7551.6 2496 236.5 356.1 1334.1 7490.8 6207.5 2397.9 589.9 602.7 2090.3 5440.5 4958.4
CCA1 3393.6 1162 215.5 105.5 477.1 2199.3 2523.8 1233 352.3 277 802.6 2103.8 1980.7
PRR9 224.8 547.8 142.3 99.7 101.5 110.2 340.9 455.7 104.2 155.7 109.1 226.1 335.1
以下のようなExcelファイルを用意して、「名前をつけて保存」で、「タブ区切りテキスト」もしくは「csv」形式で保存してください。
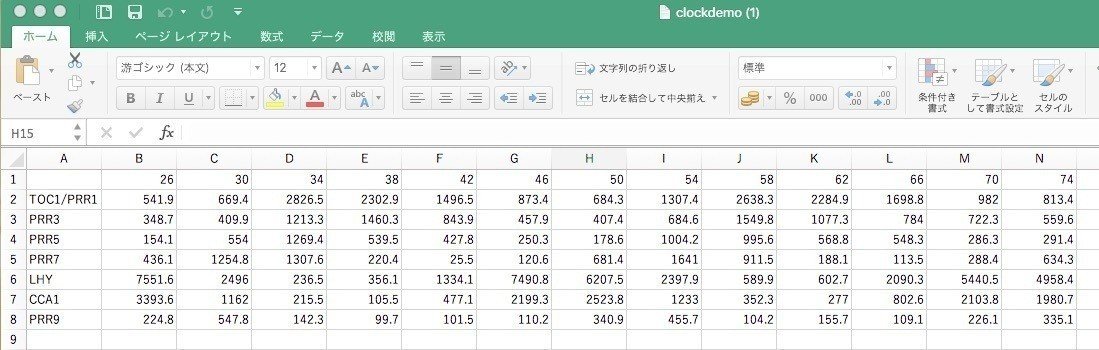
フォーマットさえ揃えれば、大きな .txt や .csvファイルでもヒートマップを描けます。
では、Rにデータを読み込みます。
> data <- read.delim("clockdemo.txt", header=TRUE, row.names=1)csvファイルの場合には、
> data<-read.csv("clockdemo.csv", header = TRUE, row.names = 1)* clockdemo.txtファイルを一旦Excelなどの表計算ソフトで開いて、csv形式で保存すると、clockdemo.csvファイルができます。
> heatmap(as.matrix(data), margin=c(4,8), main="Heat Map 1 (Raw Data)")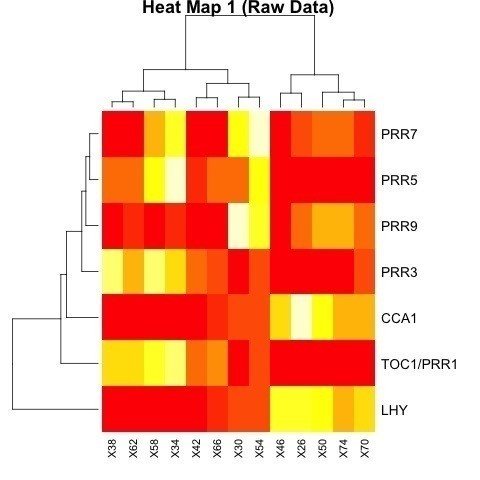
Z-score化したい場合、
> data.z <- genescale(data, axis=1, method="Z")
> heatmap(as.matrix(data.z), margin=c(4,8), main="Heat Map 2 (Z score Data)")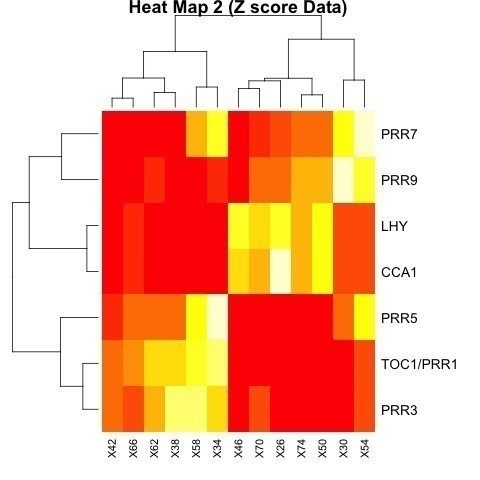
いわゆる、赤緑の図。
Time course 実験なので、X軸は並べ替えないようにパラメータを設定します。パラメータは、Colv=NA
> heatmap.2(as.matrix(data.z), col=greenred(75), scale="none", key=TRUE, symkey=FALSE, density.info="none", trace="none", cexRow=1, margin=c(4,8), main="Heat Map 2 (Z score Data)", Colv=NA)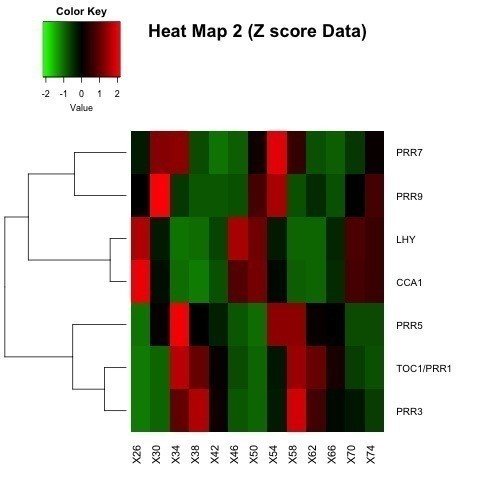
最近は、色覚異常のバリアフリー(ユニバーサルデザイン)のルールがあるので、シアンマゼンダで表示しましょう。
パラメーターは、col=cm.colors(100)
> heatmap.2(as.matrix(data.z), col=cm.colors(100), scale="none", key=TRUE, symkey=FALSE, density.info="none", trace="none", cexRow=1, margin=c(4,8), main="Heat Map 2 (Z score Data)", Colv=NA)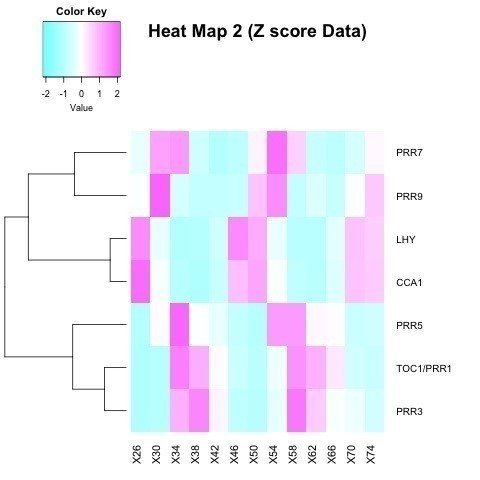
この図を見ると、LHYとCCA1が発現した後に、PRR9、PRR5、PRR3とTOC1の順番に発現していることがわかります。
ちなみに、tiffファイルとして出力する場合には、
> tiff("heatmap.tiff")
> heatmap.2(as.matrix(data.z), col=cm.colors(100), scale="none", key=TRUE, symkey=FALSE, density.info="none", trace="none", cexRow=1, margin=c(4,8), main="Heat Map 2 (Z score Data)", Colv=NA)
> dev.off()でOK。
追記
2024年6月時点でのBioconductorのインストール(https://bioconductor.org)
> if (!require("BiocManager", quietly = TRUE))
+ install.packages("BiocManager")CRANのミラーサイトを選択するように言われるので、Japanのサイトを選ぶ。
BiocManager::install(version = "3.19")コアパッケージのインストール
BiocManager::install(c("GenomicFeatures", "AnnotationDbi"))genefilterのインストール
BiocManager::install("genefilter")gplotsのインストール
> BiocManager::install("gplots")あとは、library( )でgenefilterとgplotsを読み込めば、これまでと同様にheatmapが描けるはずです。
> library(genefilter)
> library(gplots)