
PtWAVE 利用マニュアル

Version: 1.1
PtWAVEとは?
PtWAVEは、サンガーシーケンスデータ(.ab1ファイル)から、ゲノム編集効率、インデル(欠失・挿入)分布、およびそれらのアラインメントを推定するTIDE(Tracking of Indels by Decomposition)解析ツールです。従来ツールよりも50 bp以上の大きな欠失の検出に強いことが確認されており[1]、より信頼性の高いゲノム編集効率推定値が期待できます。
PtWAVEはウェブツールとして実装されており、リンク(https://www.ptwave-ptbio.com)からアクセスが可能です。 本公開バージョンは商用問わず無償で提供しております。
References
1. Nakamae et al., PtWAVE: A High-Sensitive deconvolution software of sequencing trace for the Detection of Large Indels in Genome Editing. bioRxiv. 2024.
URL:https://doi.org/10.1101/2024.04.17.589649
紹介動画
TogoTV様より操作説明に関するご紹介をいただきました。
https://togotv.dbcls.jp/20240614.html
画面モードの切り替え
ツール画面上部のタブをクリックすることで画面モードを切り替えることができます。 それぞれの画面モードでは下記のコンテンツを提供しています。

A. Aboutタブ: PtWAVEの機能解説およびポリシー
B. Input parametersタブ: データ入力画面
C. Outputタブ: 解析データ確認画面
データ入力画面の解説
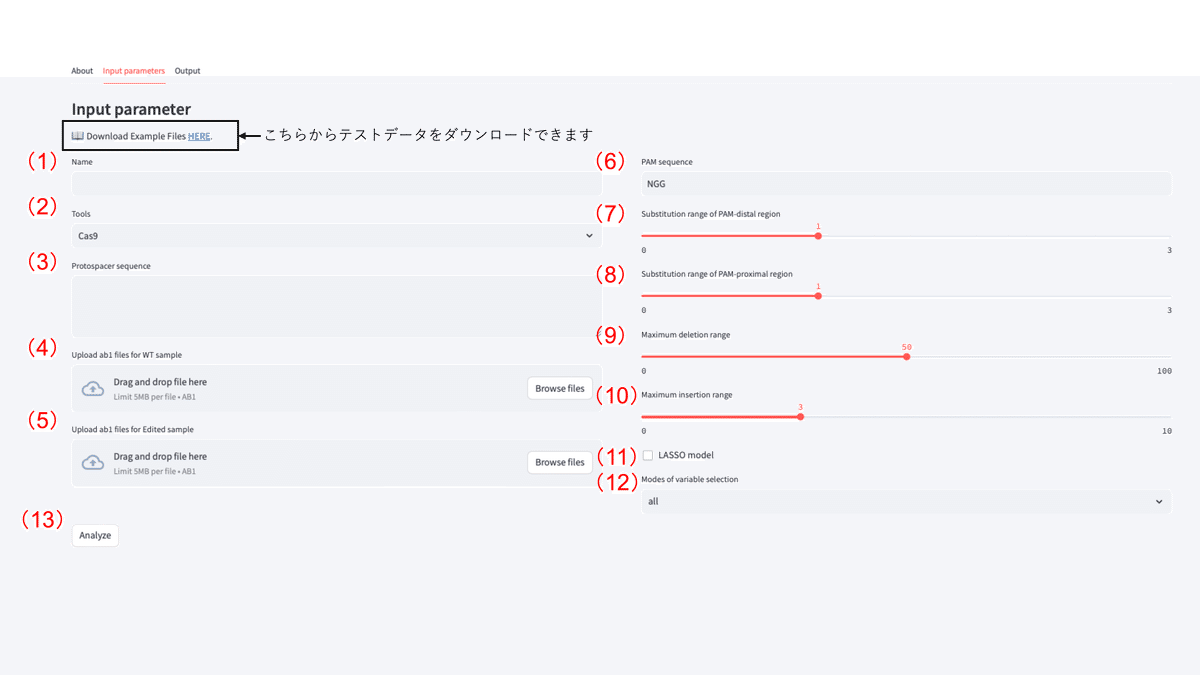
サンプル名(任意)
ツール名: ゲノム編集ツールを選択します。
※本バージョンではCas9にのみ正式対応しています。Cas12aやTALENs、ZFNs等での利用方法については「Aboutタブ」をご参照ください。プロトスペーサー: PAM配列を除く認識DNA配列(プロトスペーサー部分)を入力します。
未編集サンプルのサンガーシーケンスファイル: 編集サンプルと同じ標的領域・プライマーで未編集サンプルをシーケンスした データ(.ab1ファイル)を入力します。
編集サンプルのサンガーシーケンスファイル: 編集サンプルをシーケンスしたデータ(.ab1ファイル)を入力します。
サンプルは変異アレルタイプが10種以上想定されるBulk populationサンプルでも、数種のアレルで構成されるClonal population サンプルでも問題ありません。
シーケンスの開始地点はゲノム編集の認識DNA配列から少なくとも200 nt離れていることが望ましいです。
200bp以下、あるいは700bp以上のシーケンスデータはPtWAVEに適合しない可能性があります。
PAM配列: PAM配列を指定します。指定しない場合は"NNN"とご入力ください。
PAM遠位方向の推定置換範囲: PtWAVEが推定する切断点(Cas9ではPAMの5 '側3 nt上流)からPAM遠位方向(Cas9では5 '側)の置換変異の範囲を指定します。 置換の推定では多くの計算が伴うため、ツールの挙動が不安定になる可能性があります。本バージョンでは置換変異の存在が明確でない場合は"1 bp"で解析することを推奨しております。
PAM近位方向の推定置換範囲: PtWAVEが推定する切断点(Cas9ではPAMの5 '側3 nt上流)からPAM近位方向(Cas9では3 '側)の置換変異の範囲を指定します。 置換の推定では多くの計算が伴うため、ツールの挙動が不安定になる可能性があります。本バージョンでは置換変異の存在が明確でない場合は"1 bp"で解析することを推奨しております。
最大欠失範囲: PtWAVEが推定する切断点(Cas9ではPAMの5 '側3 nt上流)から両方向の欠失範囲を指定します。
切断点から両方向の範囲を指定するため、例えば50 bpを指定した場合は最大2×50=100 bpの欠失までを検出することができま す。
検出範囲を⻑くするほど要求されるシーケンスデータの⻑さおよび品質の水準が上昇します。エラーが生じてしまう場合はより 短い条件での実施もご検討ください。
最大挿入⻑: PtWAVEが推定する挿入変異のサイズ上限を指定します。
LASSO modelの有効化: 推定プロセスでLeast Absolute Shrinkage and Selection Operator(LASSO)を適用します。
実験的なパラメータのため基本的にはOFFを推奨しています。本機能に関する詳細な検証結果は論文をご参照ください。
変異アレルの選択アルゴリズム: PtWAVEではベイズ情報量基準 (BIC)を指標とした推定変異配列パターン(EMSP)の選択アルゴリズムが搭載しており、様々なデータに対して柔軟に高精度・高信頼性な推定結果が期待されます。本バージョンでは以下の3 種類で利用可能です。
all: 選択を行わず推定可能な全ての変異アレルを考慮します。
Clonal populationサンプルはこちらのアルゴリズムを選択することを推奨します。random: ランダムなEMSPの選択を数回実施し、BICが最も小さくなるアレルパターンを決定します。 実験的なパラメータのため基本的にはOFFを推奨しています。本機能に関する詳細な検証結果は論文をご参照ください。
backstep: 再帰的にEMSPのフィルタリングを実施し、BICが最も小さくなるアレルパターンを決定します。
Bulk populationサンプルのような複雑かつ多様なアレルパターンを含むサンプルの場合はこちらのアルゴリズムを選択することを推奨します。
解析開始ボタン: 全てのデータ入力が終了してクリックするとPtWAVEサーバー内で解析が開始されます。

クリック後にしばらく待機し、"Analysis Complete!!!"と表示されれば解析完了です。 Outputタブをクリックすることで解析結果が閲覧できます。
解析データ確認画面の解説
Figures

I. Discord plot: 提供されたデータのシーケンスシグナルの比較です。 非編集サンプル(⻘線)と編集サンプル(オレンジ線)の配列データの塩基座標(横軸)とシグナル量(縦軸)を示しています。 aln_startとaln_endの間の領域はAlignment windowと呼ばれ、解析に含まれる領域を示します。inf_startとinf_endの間の領域は Decomposition window(またはInterference window)と呼ばれ、フィッティング処理が行われた領域を示します。
基本的なゲノム編集では切断部位を中心に変異が起こるため、シーケンスデータ内においては切断部位の塩基座標から⻘線とオレンジ 線の不一致の度合いが大きくなる傾向があります。不一致が大きくなった座標が、配列データ上の実際の切断部位の塩基座標とほぼ一致すれば、解析が適切に行われたと判断可能です。一方、座標に100bp程度の大きな差がある場合は、解析に失敗した可能性がありますので入力データを再検討する必要があります。
II. Editing outcomes: 推定されたインデルの分布です。検出されたインデルの頻度(縦軸[%])をインデルのサイズ(横軸[bp])でプロットしています。未編集配列と置換配列はすべて0 bpとして集計します。
Editing efficiency

編集効率を含む数値データを示します。
III. editing_eff: 検出された変異の割合[%]。
IV. BIC: BICは解析の信頼性の指標です。値が低いほど解析の信頼性が高いとみなされます。相対的な定量指標であるため絶対的な閾値はありません。一般的に変数選択のallモードよりもbackstepやrandomモードではこの値が低くなります。
V. r_sq (R2): 変異推定の精度を表す指標です。この値が0.8以上であれば良好な変異推定結果とみなすことができます。
Sequence alignment

VI. Sequence alignment: 未編集サンプルと編集サンプルの配列データの各座標におけるプライマリーピークの塩基配列のアライメン トです。
未編集サンプルの配列に多くのインデルが観測された場合は、入力設定または配列データが不適切であることを示している可能性があります。
"Sequence alignment"セクションで提示される編集サンプルの配列はプライマリーピークの塩基配列であり推定変異配列ではない点にご注意ください。推定変異配列は"Allele contribution(normalized)"セクションで提示されます。
Allele contribution(normalized)

VII. Allele contribution(normalized): 非編集配列と推定された複数の変異配列のマルチプルアライメント結果と各変異の推定割合を示します。
Contribution: 編集サンプルにおける相対寄与率(変異頻度の推定割合)。
Mutation: 変異の種類を示すラベル。負の数は欠失、正の数は挿入、「Sub 」は置換を示します。角括弧[]は認識配列の通し番号です。この番号は変異を引き起こしたと考えられるゲノム編集ツールの認識配列番号に対応します。
本バージョンでは、認識配列は1種類しか指定を許容していないため、常に[g1]という表記になります。
Sequence: 非編集配列と推定された複数の変異配列のマルチプルアライメント結果です。「-」はギャップを表し、「|」は切断部位を表します。PtWAVEによって検出された挿入は切断部位の近傍のみ考慮されており、「n」という表記になります。
各種ポリシー・その他
ウェブサイトの著作権
本ウェブサイトの著作権は、プラチナバイオ株式会社に帰属します。
データ利用に関する規定
当サイトに入力されたデータおよび解析結果に関する権利は、すべて利用者に帰属します。 プラチナバイオ株式会社およびその関係者は、利用者の許可なく入力データおよび解析結果を他の目的で使用することを禁止し、解析終了後、入力データおよび解析結果を直ちにサーバーから削除します。
利用ライセンス
本ウェブサイトで公開されているPtWAVEは、許諾なく非営利・営利の両方の目的でご利用いただけます。
お問い合わせ
PtWAVEに関する技術的なお問い合わせは下記のアドレスまでご連絡ください。
広島大学 ゲノム編集イノベーションセンター PtBio共同研究講座 バイオDX研究室中前 和恭
e-mail: kazuki-nakamae<アットマーク>hiroshima-u.ac.jp
PtWAVEのサービス・運営に関するお問い合わせはプラチナバイオ株式会社の問い合わせ窓口までご連絡ください。
プラチナバイオ株式会社
https://www.pt-bio.com/
開発チーム
解析エンジン: 伊出 佐耶・中前 和恭
ウェブインターフェース: 大貫 永輝・中前 和恭
マニュアル: 中前 和恭
監修: 小野 浩雅・石井 敦浩・奥原 啓輔