タンパク質-タンパク質界面における共進化が、de novo構造予測によるオリゴマータンパク質複合体の化学量論的推論を導く

記事内容へスキップ
記事情報へスキップ
ワイリーオンラインライブラリー
分子微生物学早わかり
リソース
オープンアクセス
タンパク質-タンパク質界面における共進化が、de novo構造予測によるオリゴマータンパク質複合体の化学量論的推論を導く
https://onlinelibrary.wiley.com/doi/full/10.1111/mmi.15169

マックス・キリアン, イルカ・B・ビショフス
初出:2023年9月30日
https://doi.org/10.1111/mmi.15169
論文概要
セクション
要旨
特異的な化学量論に基づく四次構造は、タンパク質複合体の特異的機能にとって極めて重要である。しかし、多くのタンパク質複合体の構造を実験的に決定することは、依然として大きなボトルネックとなっている。深層学習アルゴリズムAlphafold2-multimer(AF2-multimer)のような構造バイオインフォマティクスアプローチは、正確なde novo構造と接触予測のために、アミノ酸と配列-構造の共進化を活用する。擬似尤度最大化直接結合解析(plmDCA)は、統計的モデリングによって共進化する残基ペアを検出するために用いられてきた。ここでは、この2つの手法を組み合わせることで、タンパク質複合体の4次構造と化学量論のde novo予測に利用できることを示す。我々は、既存のAF2-多量体信頼性評価指標を解釈可能なスコアで補強することにより、分子間界面における共進化残基対のネイティブコンタクトの最適な割合を持つ複合体を同定する。この手法を用いて、構造未知の枯草菌胞子発芽タンパク質複合体の4次構造と非自明な化学量論を予測した。したがって、分子間界面における共進化は、構造未解明の細菌タンパク質複合体のAIベースのde novo四次構造予測と相乗効果をもたらす可能性がある。
1 はじめに
タンパク質と複雑な分子機械との結合は、代謝からシグナル伝達まで、生命のあらゆる領域にわたる多くの細胞内プロセス(Alberts, 1998)の鍵となる(Drew et al., 2021; Gavin et al., 2006; Krogan et al., 2006; Kühner et al., 2009; Luck et al.) 例えば、対称的複合体へのホモオリゴマー化は、タンパク質の安定性を高め、サブユニット間のアロステリック制御を可能にし、ゲノムサイズを増大させることなくモジュラー複合体のアセンブリを促進することができる(Bergendahl & Marsh, 2017; Goodsell & Olson, 2000; Hashimoto et al.) その延長として、ヘテロオリゴマー化は、異なるタンパク質の異なる機能を空間的に近接させることによって結合させ、それによって制御経路と代謝経路の両方の特定の構成要素を接続する(Marsh et al.) 構造的に特徴づけられたヘテロオリゴマー複合体は、一般的に自明でない不均一な化学量論を持っており(Ahnertら、2015;Marsh & Teichmann、2015)、タンパク質複合体の4次構造が生体内での特異的機能の鍵であることを示唆している(Ahnertら、2015;Cai、2008;Gaber & Pavšič、2021;Kimら、2019;Marshら、2015;Marsh & Teichmann、2015)。複合体組成とその高分子4次構造は、複合体を精製した後、生化学的、生物物理学的、または構造生物学的手法によって実験的に同定することができる(Gaber & Pavšič, 2021; Marsh & Teichmann, 2015; Nealon et al. 例えば、膜タンパク質の構造は、膜貫通シグナル伝達(Lefkowitz, 2007; Westerfield & Barrera, 2020; Zschiedrich et al、 2009; Roche & Törnroth-Horsefield, 2017)、異種発現や精製が困難な性質のため(Hendrickson, 2016)、Protein Data Bank (PDB)にはあまり掲載されていないが、プロテオームの20~30%を占めると推定されている(Wallin & Von Heijne, 1998)。さらに、PDBで構造決定されたタンパク質の大部分はモノマーまたはホモオリゴマーであるが、冗長でないヘテロオリゴマー構造は、比較するとまだ十分に知られていない(Marsh & Teichmann, 2015)。そのため、このような複合体のin vivo研究は、例えば部位特異的突然変異誘発実験などの構造情報の不足がボトルネックとなっている。
近年、ディープラーニングに基づく構造バイオインフォマティクスの手法が、共進化情報とともに配列と構造の関係を計算によって利用することで、タンパク質の原子構造をde novoで予測することに大きな進歩をもたらしている(Baek et al.) これらのうち、Alphafold2(AF2)とAF2-multimerは、それぞれ配列のみから単量体とオリゴマーの複合体構造を高精度に予測する(Bryant, Pozzati, & Elofsson, 2022; Evans et al.) これらの予測は、定期的に実験的検証と一致することが示されている(Evansら、2022; Hegedűsら、2022; Jumperら、2021; Leeら、2022; Stevens & He、2022; Tunyasuvunakoolら、2021; Yinら、2022)。しかし、AF2-multimerは、タンパク質入力中の多量体を自動的に検出することができないため、配列のみからタンパク質複合体の4次構造を決定するためのトレーニングは行っていない。AF2-multimerは、複合体に折り畳まれるタンパク質鎖の化学量論をユーザーが入力する必要がある(Bryant, Pozzati, Zhu, et al.) さらに、AF2-multimerの信頼性指標は、化学量論が既知の複合体予測でのみベンチマークされている(Evans et al., 2022; Yin et al.) これらの指標はまた、AF2-multimerネットワークがある化学量論で特定の(確信度の高い)複合体予測を生成した理由をほとんど示していない。最後に、AF2-multimerネットワークが単独で、タンパク質複合体の正しい化学量論と四次構造を同定する予測力を持つかどうかは示されていない。AF2-およびAF2-多量体ベースの手法は、タンパク質複合体の構造的に分解された相互作用を予測するために、生命科学分野で急速に採用されている(Burke et al.
ここでは、共進化解析、特に擬似尤度最大化直接結合解析(plmDCA)(Ekeberg et al. これらのMSAにおけるアミノ酸ペアの共変化は、統計物理学に由来する確率論的最大エントロピーモデルに従ってカップリングパラメータを推論することによって検出される(Balakrishnan et al.) タンパク質のインターフェースは進化の過程で同族パートナーとの相互作用を維持するため(Aakre et al., 2015; Ovchinnikov et al., 2014)、空間的に近接するアミノ酸は共進化し、その結果、進化を通じてタンパク質のフォールドと相互作用インターフェースを維持する変異が共変化する(Deane & de Oliveira, 2017; Göbel et al., 1994)。タンパク質配列に対するこれらの進化的制約は、タンパク質-タンパク質界面においても残基近接性の予測因子(Hopf et al., 2012; Kamisetty et al., 2013; Marks et al., 2011; Morcos et al., 2011; Ovchinnikov et al., 2015)を提供し、したがって保存されたタンパク質複合体の構造に関する情報を含む(Anishchenko et al., 2017; Cong et al., 2019; Green et al., 2021; Hopf et al., 2014)。先行研究はまた、タンパク質の三次構造で離れた共進化残基対は、タンパク質-タンパク質界面での鎖間接触でもあり、それによってタンパク質複合体の四次構造に寄与することを示している(Anishchenkoら、2017; Hopfら、2012; Marksら、2011)。
そこで、de novoタンパク質構造予測と共進化解析の統合的アプローチにより、オリゴマー化界面での共進化を同定することで、タンパク質複合体の進化的に保存された4次構造を計算機で推測することができると考えた。
本研究では、Alphafold2-multimerによるde novo構造・接触予測と、擬似尤度最大化直接結合解析を組み合わせることで、細菌タンパク質複合体の分子間界面における共進化を追跡する。我々は、実験的によく特徴付けられた酵素複合体でこのアプローチをテストし、次に枯草菌の胞子発芽タンパク質複合体に我々の戦略を適用した。これらの複合体は、実験的にも構造的にも特徴づけが難しいことで有名であるが(Christie & Setlow, 2020)、自明でない化学量論で複合体を形成することが予測される。我々のデータに基づくと、Alphafold2-多量体信頼性指標と界面の共進化解析が相乗効果を発揮して、タンパク質複合体の化学量論的構造を推定することが示唆される。このように、我々のアプローチは、分子間界面における共進化を追跡することで、Alphafold2-多量体予測の解釈可能性を高め、AIを用いたタンパク質複合体のde novo四次構造予測の指針となる。
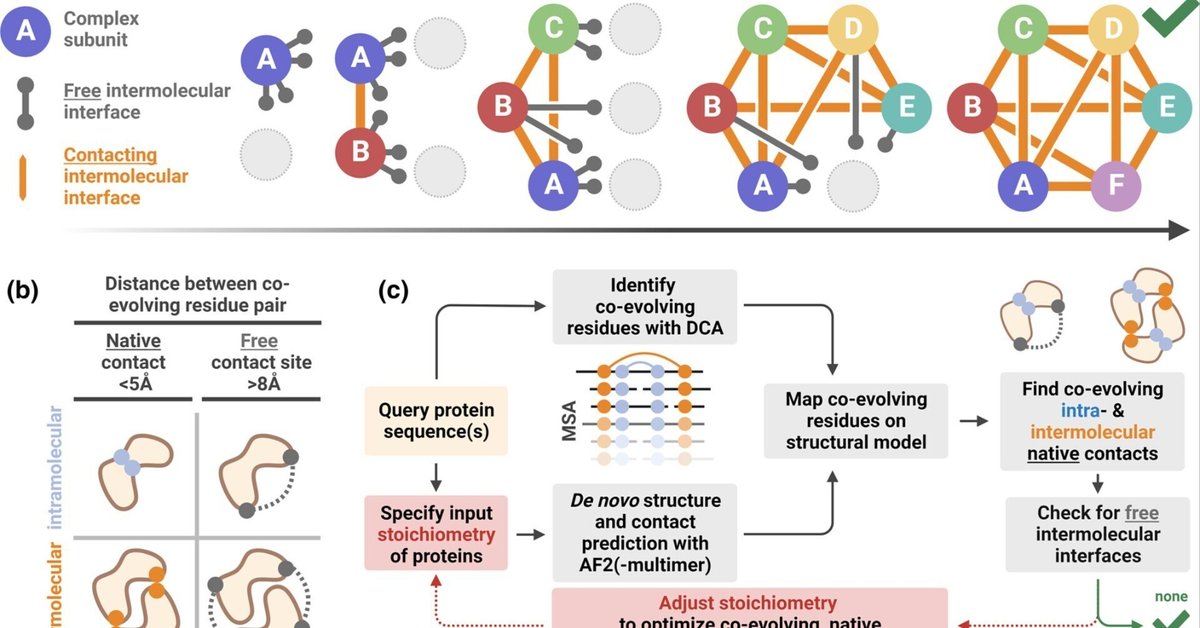
2 モデルとワークフロー
特定の四次構造と化学量論を持つ機能的に活性なオリゴマー蛋白質複合体の組み立ては、分子間の蛋白質-蛋白質相互作用によって駆動される。フリーな分子間インターフェースは、追加サブユニットと相互作用し、すべての分子間インターフェースがかみ合うまで、高次オリゴマーのアセンブリを促進する(Ahnert et al. 化学量論が正しい完全な複合体では、これらのオリゴマー化界面はこのように「飽和」しており、これらの界面にあるアミノ酸対は同族パートナーと直接物理的に接触している。タンパク質(複合体)の構造モデルにおいて、空間的に近接しているこれらのアミノ酸対を、以下ではネイティブコンタクトと呼ぶ(Méndez et al.) そこで我々は、AF2-multimerによって予測された構造モデルの界面にある5Å未満のネイティブコンタクト(Anishchenko et al., 2017; Méndez et al., 2003)に注目することで、タンパク質複合体のde novo四次構造予測を導くことができるのではないかと考えた。これらの予測された分子間ネイティブコンタクトのどれが、plmDCA(以下、単にDCAと呼ぶ)によって同定された共進化するアミノ酸対に対応するかを同定し、複合体のタンパク質-タンパク質界面における共進化を追跡する(図1b)。
詳細は画像に続くキャプションを参照。
図1
図ビューアーで開く
パワーポイント
キャプション
これらの共進化残基は、構造的に類似した複合体において保存された機能を共有している可能性が高い、十分に分岐しているが相同なタンパク質配列を含むMSAにおいて同定される。ヘテロオリゴマーにおけるインターフェースの共進化を解析するために、配列はゲノム近傍にある(保存されたオペロンにおける遺伝子の共局在化)か、相互ベストヒット(クエリに対してパラログではなくオルソログである)のどちらかによって、種ごとのMSAでもペアになっている(Cong et al.) 遺伝的ドリフトにもかかわらずタンパク質の機能と(複雑な)構造を維持する選択圧があるため、より深いMSAでより分岐した配列は、一般的に共進化のより強い証拠を提供する(Avila-Herrera & Pollard, 2015; Green et al.) したがって、配列同一性が高い冗長配列(ここではArtzi et al., 2021; Gao, Barajas-Ornelas, et al., 2022と同様に90%以上)は、DCA中にクラスタ化され、まとめて再重み付けされ、サンプリングされた配列の系統的多様性が増加する。MSA内の系統学的に分岐の多い配列については、より低い配列同一性が選択されることもある(例えば80%以上)(Green et al., 2021; Hopf et al., 2014)。
これらの有効な非冗長配列の総数(Neff)をクエリー長(L)で割ったものが、MSAの深さと広さを表す。Neff/Lが0.3-1以上のMSAは、DCAベースの手法で共進化残基を正確に検出するのに十分であることが示されている(Hopf et al.) しかし、Neff/Lがさらに高いMSAは、一般に、分子内および分子間の共進化残基ペアを捕捉するのに優れた性能を発揮する(Avila-Herrera & Pollard, 2015; Hopf et al.) 逆に、非常に深いMSAを選択した場合の潜在的な落とし穴は、タンパク質(スーパー)ファミリー間の非常に離れたホモログにおけるインターフェースの分岐である可能性がある(Aloy et al.) この結果、共進化解析におけるMSAの深さと系統的多様性の間のトレードオフは、ケースに依存することになる。したがって、特定のタンパク質複合体を解析するためにMSAを慎重に組み立てることは、共進化解析を成功させるために極めて重要である。我々の解析では、表S1および表S2にまとめたように、DCA用の(対になった)MSAのうち、深さが中程度から高く(Neff/L≈3-30)、クエリー配列のカバレッジが高い(>80%-90%)ものを選択した。後者は、ギャップが多い断片配列やクエリーカバレッジの低い断片配列によるMSAの偽相関を避けるために重要であり、そのためMSAアセンブリー中に除去した(Methods参照)。さらに、選択したMSAで検索されたすべての配列が類似のタンパク質ファミリーに割り当てられていることを確認し、機能が乖離した非常に離れたホモログが含まれないようにした。
DCAから得られたこの共進化情報を手元に置き、次にAF2-多量体予測複合体(図1c)内の界面におけるネイティブな分子間接触に対応する共進化残基の割合を最適化する複合体化学量論を探索する。この目的のために、構造モデルを計算し、DCAによって同定されたすべての共進化残基対を予測モデル上にマッピングする。複合体のサブユニット内およびサブユニット間の最小重原子残基-残基距離を決定し、AF2-多量体モデルにおけるすべてのネイティブな分子内あるいは分子間接触を見つける。この距離閾値以下の残基対は、疎水性相互作用または水素結合を介した両アミノ酸の直接的な物理的相互作用により、実験構造において最も強く共進化することが示されている(Anishchenko et al.) 共進化する残基ペア間の大きな空間距離は、推定上の分子間界面における潜在的相互作用を示す傾向がある(Anishchenko et al.) したがって、接触していない(>8Å)共進化残基のかなりの量(Artziら、2021;Hopfら、2014;Ovchinnikovら、2014)は、非ネイティブ「フリー接触部位」と呼ばれ、複合体が不完全であることを示す。言い換えれば、対象の複合体における共進化の「パターン」は、AF2-multimerによって予測される接触とは一致しない。次に、共進化する残基対の分子間ネイティブ接触の最適値に到達し、共進化の「パターン」がAF2-multimerによって予測された接触と一致するまで、構造予測の入力化学量論を調整することによって、AF2-multimerで高次複合体を繰り返しモデル化する。
3 結果
3.1 既知の六量体酵素であるAldの四次構造の計算推論
我々のモデルを検証するために、広く構造的にも実験的にもよく特徴付けられたホモオリゴマータンパク質複合体を選んだ。したがって、枯草菌(BsAld)由来のアラニン脱水素酵素(Ald, EC 1.4.1.1)を選択した。この代謝酵素は、補酵素NAD+を用いてL-アラニンをピルビン酸とアンモニアに可逆的に脱アミノ化する反応を触媒する(Grimshaw & Cleland, 1981; Wiame & Piérard, 1955)。Aldは歴史的に、発芽栄養素L-アラニンに応答する細菌内胞子の発芽受容体であるという仮説が立てられており(Freeseら、1964;O'Connor & Halvorson、1961)、Aldは発芽には必要ではないが、枯草菌内胞子の発育(Siranosianら、1993)および胞子復活の後期段階において重要な役割を果たし、うまく伸長する(Mutluら、2018、2020)。
現在のところ、BsAldの完全な実験的構造は得られていない。しかし、Bacillus cereus、Geobacillus stearothermophilus、Lysinibacillus sphaericusなど、近縁の他のBacilli由来のAldは、ホモヘキサマーを形成することが実験的に示されており(Kuroda et al., 1990; Porumb et al., 1987)、結核菌からは、基質が結合したAldと結合していないAldのX線結晶構造が入手可能である(Ågren et al., 2008; Tripathi & Ramachandran, 2008)。これらの実験構造では、Aldは3回二面体対称性を持つホモ六量体複合体に配列しており、シアノバクテリウムPhormidium lapideum(Baker et al., 1998)とグラム陰性好熱菌Thermus thermophilus(PDB: 2EEZ)のX線結晶構造も非常によく似た構造とトポロジーを共有している。従って、細菌アルドホモログの個々の鎖がホモ六量体複合体に集合することは、進化的に広く保存されている。
3.1.1 AF2-多量体による化学量論が異なるBsAld複合体のモデリング
相同なホモ六量体 BsAld 構造を推定するために、AF2 を用いてモノマーから開始し、AF2-multimer 構造予測ごとに合計 8 鎖まで BsAld 鎖を 1 つずつ追加していき、不偏的な方法で BsAld 鎖の数を増やしたモデルを構築し(図 2a)、それぞれのケースで上位にランクされたモデルの信頼性指標を比較した(図 2b,c)。AF2-multimerは、TM-score similarity metric (Xu & Zhang, 2010; Zhang & Skolnick, 2004)に基づき、サブユニットフォールドの信頼性をスコア化するための予測テンプレートモデリングスコア(pTM, 0〜1)と、複合体構造の信頼性をスコア化するためのインターフェースpTM(ipTM, 0〜1)を生成する。単量体でさえもpTMが0.9を超え、AF2およびAF2-多量体予測(Jumper et al. ipTM>0.85の予測における分子間界面もまた、実験的検証とほぼ一致している(O'Reilly et al., 2023)。一般的にpTMとipTMのスコアの差は、高信頼性領域でもかなり異なることがあることに注意されたい(Evans et al., 2022; Jumper et al., 2021)。しかし、化学量論が正しい6量体予測は、pTMとipTMのスコアが最も高かった。また、ほとんどの構造予測で低い予測整列誤差(PAE、0~31.75Å)が観察され(図2c)、ほぼすべてのBsAld複合体構造予測において、個々の残基とドメインの相対的な位置に対するAF2-multimerの信頼性が高いことが示された(Varadi et al.) さらに、BsAldモノマーの平均残基当たり予測局所距離差検定(pLDDT、0~100)スコア(95.6±8.9)は、6量体複合体予測(93.0±9.4)と同程度であり、いずれの構造モデルの平均残基当たり精度も高い信頼性を示している(Jumper et al.) したがって、これらすべてのAF2-多量体信頼性指標のばらつきが、真の「シグナル」なのか、単なるノイズなのかは不明であり、BsAldの正しいオリゴマー状態はすぐには明らかにならない。
詳細は画像に続くキャプションを参照。
図2
図ビューアーで開く
パワーポイント
キャプション
3.1.2 BsAldの単量体AF2モデルにおけるホモオリゴマー化界面のDCAによる同定
次に、BsAldホモログのMSAにDCAを適用して、BsAld内で有意に共進化している残基対を推定した。 有効配列総数11,234、Neff/L = 30.5のアラインメントを選択した。このアラインメントは、細菌門(Bacillota、Bacteroidota、Actinomycetota、Pseudomonadota、Cyanobacteriotaなど)の広い範囲をカバーしており、事実上すべての配列がAldファミリーに属すると注釈されている。より深いアラインメントも作成されたが、バクテリア、古細菌、真核生物から多くの離れたAldホモログが含まれており、そのほとんどがAldとは異なる機能を持つことが注釈されていたため、解析には選ばれなかった。
選ばれたMSAの2残基間のDCAによって決定された共進化の「程度」は、補正ノルムスコア(cn-score)によって表された(Ekeberg et al.) この指標は、配列共進化の大域的確率モデル(Balakrishnan et al., 2011; Ekeberg et al., 2013)に従って、MSAにおける残基-残基結合の大きさを定量化する。先に述べたように(Hopf et al., 2014; Toth-Petroczy et al., 2016)、タンパク質配列中で離れているほとんどの残基対は共進化せず、高得点の外れ値のみが空間的に近接した強く結合した共進化残基を表す。そこで、cn-score分布(Toth-Petroczy et al., 2016)の高スコアテールにある事後確率0.9以上の残基対で、一次配列でも少なくとも6アミノ酸離れているものだけを共進化残基として選択した(図S1a、Methods参照)。長さLのタンパク質は、L!/2×(L-2)を持つことができる!(すなわち、Lは2を選ぶ)共進化しうるユニークなアミノ酸対を持つことができる。378個のアミノ酸を持つBsAldの場合、これは71,253個のユニークなペアに相当する。われわれのDCAの基準では、これらの残基ペアのうち共進化が確認されたのはわずか552ペア(0.77%)であった。しかし、BsAldの325アミノ酸(86%)がこれらのペアに寄与している。このように、共進化する残基対は比較的少ないが、実質的にタンパク質全体に広く分布している。
次に、これらの共進化残基対が、AF2およびAF2-多量体構造モデルにおけるネイティブコンタクトに対応するかどうかを評価した。単量体BsAld AF2モデルでは、共進化残基対の68.7%が5Åより近接しており、構造予測における分子内ネイティブコンタクトに相当した。しかし、共進化残基のかなりの部分は、非ネイティブな自由接触部位として8Å以上離れたままであった(図S1b)。これらの共進化残基対の上位のいくつかはタンパク質の周辺領域にマップされ、ホモオリゴマー化を促進する分子間界面に位置している可能性が示唆された(図3a,b)。実際、AF2-multimerで作成した六量体構造モデル(図S1c)では、これらの共進化する残基対のほとんどすべてが近接しており、ホモ六量体BsAldモデルの溶媒を除いた分子間界面で5Å以下のネイティブコンタクトに対応していた(図3c,d)。これらの推測されたホモオリゴマー界面は、結核菌のAld(Ågrenら、2008)(PDB: 2VHZ、図S2a-d)内の実験構造および共進化残基とも一致している。AF2およびAF2-multimerによるde novo構造予測とDCAによる共進化解析を組み合わせることで、オリゴマー化界面を突き止めることができる。
詳細は画像に続くキャプションを参照。
図3
図ビューアーで開く
パワーポイント
キャプション
3.1.3 BsAldホモ六量体における共進化するネイティブな分子間接触の最適化
次に、AF2-多量体複合体予測において、ネイティブな分子間接触に相当する共進化残基対の割合が、BsAldサブユニットの数によってどのように変化するかを解析した。分子間タンパク質相互作用界面におけるこれらの共進化するネイティブな接触の割合は徐々に増加し、BsAldサブユニット6個で最適に達した(図3e)。BsAldサブユニットをさらに追加しても、構造予測においてこれらの共進化するネイティブコンタクトの割合が増加することは見いだされなかった。なぜなら、対称的複合体において、界面でのネイティブコンタクトを維持したまま、さらにプロトマーを追加することができるからである。したがって、6つのBsAldサブユニットの相互作用は、BsAld複合体の分子間界面のほとんどの共進化残基を特異的に飽和させるのに十分であると考えられる。さらに、BsAldサブユニットの数が増えても、ネイティブな分子内コンタクトの割合はほとんど変化しなかった(すべての予測で67.7%-69.0%)ことから、AF2-multimerはすべての複合体予測において、BsAldサブユニットに対して一貫したフォールドを予測していることが示された。言い換えれば、BsAldにコードされている共進化の「パターン」は、六量体構造予測においてAF2-multimerが予測した接触と最もよく一致している。BsAldの六量体AF2-多量体構造予測は、他のすべての化学量論と比較して、pTMとipTMのスコアも最も高かった(図2b)。この予測された構造は、ホモログにおける広範な先行実験結果(Ågrenら, 2008; Bakerら, 1998; Kurodaら, 1990; Porumbら、 1987; Tripathi & Ramachandran, 2008)、さらに閉じたコンフォメーションにおけるM. tuberculosis holo-Ald構造(Ågren et al., 2008)と高い構造類似性を示している(PDB: 2VHZ, TM-score: 0.81, Cα-RMSD: 2.03Å)。
3.2 二量体の二量体として知られるピルビン酸脱水素酵素複合体のE1モジュールの四次構造の計算推論
ペアMSAの共進化解析(Green et al., 2021; Hopf et al., 2014; Ovchinnikov et al., 2014)とAF2を用いたde novo構造モデリング(Bryant, Pozzati, & Elofsson., 2022; Burke et al., 2023; Evans et al. そこでわれわれは、構造的にも実験的にもよく特徴づけられたヘテロオリゴマーを用いて、われわれの戦略の適用性をテストしようとした。我々は、B. subtilis PdhAとPdhBを選択した。これは、大きな多酵素ピルビン酸デヒドロゲナーゼ複合体(PDHC)のE1モジュール(EC 1.2.4.1)のαサブユニットとβサブユニットに相当する。E1は補酵素であるチアミンピロリン酸でピルビン酸を脱炭酸し、PDHCによるピルビン酸からのアセチル-CoA産生の開始段階となる(Patel et al.) PDHCはこのように解糖とクエン酸サイクルを結びつけ、生命のあらゆる領域における代謝において中心的な役割を担っている。E1の構造は、グラム陽性細菌と真核生物の両方において、2つのαサブユニットとβサブユニットからなる「二量体の二量体」として広く保存されている(Neveling et al.) B. subtilis E1の構造は実験的に解明されていないが、近縁のGeobacillus stearothermophilus E1モジュールは構造的に特徴付けられており、期待される「二量体の二量体」を形成している(Frank et al.、2004)。
そこで我々は、AF2-multimerを用いてヘテロオリゴマー複合体モデルを構築し、PdhAとPdhBのペアMSAを用いて共進化解析を行うことにより、枯草菌PDHCのE1モジュールの相同構造をモデル化することを試みた。ヘテロオリゴマーには、ホモオリゴマーよりも多くのサブユニットの組み合わせがある。2つのタンパク質間の二項相互作用が実験的に確認されたとしても、ヘテロオリゴマーでは不均一な化学量論が一般的であり(Marsh et al. これをエミュレートするために、枯草菌PDHC E1モジュールのAF2-多量体予測において、個々のサブユニットの数を偏りのない方法で増やし続けた。化学量論が1:1のPdhA-PdhB複合体から始め、化学量論が均一な複合体と不均一な複合体の両方で個々のサブユニットを追加し続けた(図S3a)。AF2-multimerは1:1、1:2、2:1、2:2の化学量論のPdhA-PdhB複合体を極めて高い信頼性で予測したが、2:2複合体を超える高次オリゴマーではモデルの信頼性が急激に低下するか、AF2-multimerでは完全な単一複合体は予測されなかった(図S3b-d)。ipTMスコアは1:2および2:1複合体では0.96、2:2複合体ではipTM = 0.97であり(図S3c)、AF2-multimerは枯草菌PDHC E1モジュールの不完全な部分複合体に対しても極めて高い信頼性を与えることが示された。
次に、DCAによって同定された共進化残基ペアをこれらの予測された複合体にマッピングした。ヘテロオリゴマー複合体には、ホモオリゴマーよりも多くの共進化可能な残基対が存在する。L1とL2の長さを持つ2つのタンパク質では、L1 choose 2 + L2 choose 2のユニークで冗長でない残基ペアが存在し、どちらかのサブユニット内で共進化する可能性がある。その上、L1×L2残基対は、選択圧の下での分子間相互作用により、両タンパク質間で共進化する可能性がある。PdhAとPdhBのアミノ酸数がそれぞれ371と325の場合、これは241,860対という圧倒的な数に相当し、そのうち50%近くが潜在的なヘテロマー残基相互作用である。逆に、主に細菌由来の11,331の有効配列(Neff/L = 16.28)を用いたペアMSAのDCAによって同定された、我々の基準で共進化する残基ペアの総セットは、はるかに小さい:471と561の共進化残基ペアがそれぞれPhdAとPdhBにマップされ、両タンパク質間で共進化する残基ペアはわずか28である。言い換えると、枯草菌PDHC E1モジュール内のユニークな残基対のわずか0.44%しか共進化していない。
これらの共進化残基を予測される複合体にマッピングすると、PdhAとPdhBが1:1の複合体内および複合体間でかなりの量のフリーコンタクトが存在し、PdhAとPdhBの化学量論が不均一な複合体では、これらのフリーコンタクトの大部分はまだ残っており、複合体周辺部の反対側にマッピングされた(図4a)。この解析から、PdhAまたはPdhBはともに、サブコンプレックス中で遊離したままの広範なホモ二量化界面を持つことが強く示唆された。したがって、PdhAとPdhBの2:2複合体の溶媒で排除されたホモおよびヘテロマー界面では、事実上すべての自由接触が、共進化するネイティブな分子間接触に対応していることがわかった(図4b)。共進化解析に基づくスコアリングでも、2:2複合体の共進化するネイティブな分子間接触が、他のすべての複合体よりもわずかに改善されていることが示された(図4c)。BsAldホモオリゴマーに関しては、3:3の高次複合体では、共進化するネイティブな分子間コンタクトの改善は見られなかった。このような高次複合体はipTMスコアも2:2複合体よりかなり低く、共進化解析のスコアリングアプローチと一致した。
詳細は画像に続くキャプションを参照。
図4
図ビューアで開く
パワーポイント
キャプション
この予測された構造と推定された2:2の化学量論は、G. stearothermophilus由来のPDHC E1モジュール(PDB: 1W85, TM-score: 0.99, Cα-RMSD: 0.42Å)(Frankら, 2004)から予想される「二量体の二量体」と非常に一致している。BsAldホモオリゴマーに関しては、共進化解析に基づくスコアリングは、AF2-多量体信頼性評価指標と相乗効果を発揮し、枯草菌PDHCのE1モジュールの不完全なサブコンプレックスを同定するのに特に有用であることが証明された。
3.3 枯草菌の胞子内発芽に関与するオリゴマー複合体の予測
数十年にわたる研究にもかかわらず、構造情報が不足している興味深いシグナル伝達系は、枯草菌の胞子内発芽装置である。このグラム陽性細菌は、代謝的に休眠状態の内胞子を形成し、劣悪な環境条件に耐えることができる。特定の栄養素を感知すると、これらの胞子は急速に発芽して復活する(Higgins & Dworkin, 2012; Tan & Ramamurthi, 2014)。栄養素の発芽は、胞子内膜の発芽受容体(GR)によって感知される。枯草菌の主要なGRであるGerAは、トリシストロニックオペロンにコードされており、L-アラニンに応答して胞子の発芽を誘発する。GerAの活性化は、胞子コアからのジピコリン酸(DPA)と呼ばれる低分子の放出を促進する、ヘプタシストロニックspoVAオペロンにコードされるチャネルを開く(Setlow et al.) このチャネルの必須構成要素はspoVAC-D-EBによってコードされているが(Gao, Barajas-Ornelas, et al., 2022)、spoVAの他の遺伝子もDPAチャネルの構成要素であることが示唆されている(Setlow et al. GRの個々のサブユニットの可溶性ドメイン(Li et al., 2010, 2019)およびDPAチャネル(Li et al., 2012)のX線結晶構造は入手可能であるが、これらの複合体のいずれについても完全な実験的構造はなく、化学量論は不明である。そこで我々は、gerAオペロンとspoVAオペロンにコードされる複合体の4次構造と化学量論を予測するために、計算論的アプローチを用いた。
共進化解析のために、先行解析と同じMSAアセンブリー基準を用いて、サブユニットの(ペア)MSAをアセンブリーした。すべてのMSAについて、有効配列の総数と深さを表S1とS2に示し、その中で検出された共進化残基対の数を含む。すべてのクエリーの配列は、既知のすべての胞子形成細菌を含むBacillota門の細菌からのみ検索した(Galperin et al., 2022)。胞子発芽における特異的な機能のため、これらのタンパク質は、これまで解析してきた酵素複合体よりもはるかに狭い範囲で保存されている可能性が高い。
3.3.1 GerAのaサブユニットはホモオリゴマー複合体を形成すると予測される
GRは一般にA、B、Cと呼ばれる3つの膜結合型サブユニットから構成され(図5a)、胞子形成細菌に特有のひだを持つ(Artzi et al.) Bサブユニットには栄養結合ポケットが存在する可能性が高いことが示されているが(Artzi et al. そこで我々は、GRサブユニットが高次複合体を形成し、発芽時のシグナル伝達に関与しているかどうかを評価した。そのために、AF2-multimerを用いたde novo構造予測と、GerAの全サブユニットの共進化解析を行った。単量体構造予測において自由な接触部位を検出することで、GerAのAサブユニット(GerAA)は自由なホモオリゴマー化界面を持っている可能性があることがわかった(図5b)。GerAAは、構造的に類似した2つの可溶性N末端ドメイン(NTD1&2)が非構造化リンカー(Li et al. AF2-multimerはGerAAの高次複合体を確実に予測し(図S4a-d)、環状対称性を有するGerAA五量体または六量体において、共進化するネイティブな分子間接触の最適値に達することを見出した(図5c)。6量体では、5量体よりもわずかにネイティブな共進化分子間接触が多いが、ipTMは低い。AF2-多量体の信頼性、あるいは共進化に基づくスコアリング手法のいずれかに基づけば、GerAAはいずれのオリゴマー状態も同様に妥当である。
詳細は画像に続くキャプションを参照。
図5
図ビューアーで開く
パワーポイント
キャプション
GerAAホモ6量体では、これらの共進化するネイティブな分子間接触は、隣接するGerAAサブユニット間の広範な界面にマッピングされ、その接触面積は〜3400Å2と大きい。隣接するNTD2ドメインは互いに、またそれぞれのCTDと強固に相互作用し、隣接するサブユニットのTMドメインは広範囲に連動する(図6a)。これらの予測される相互作用は、生体内ではGerAAの接触面上の疎水性パッチと極性パッチの両方の相互作用によって媒介される可能性があり(図S5a)、GerAのシグナル伝達特性を変化させる以前に特徴付けられた変異(Amonら、2022;Grelaら、2018;Mongkolthanarukら、2011)は、これらのホモオリゴマー界面にマッピングされる(図S5b)。重要なことに、異なる栄養特異性を持つGRの一部である枯草菌のGerAAのパラログ、GerBAとGerKAも同様に、ホモ六量体複合体を形成すると確信をもって予測された(図S5c,d)。したがって、GRのAサブユニットのホモオリゴマー化は、このタンパク質ファミリーに保存された特徴である可能性が非常に高い。実際、ある独立したグループは、GerAAがホモオリゴマーを形成し、栄養ゲートイオンチャネルとして機能する可能性があることを生化学的およびin vivoで確認している(Gao et al.)
詳細は画像に続くキャプションを参照
図6
図ビューアで開く
パワーポイント
キャプション
3.3.2 SpoVAチャネルはヘテロ三量体の二量体を形成すると推測される
SpoVAC-D-EBによって形成されるチャネルは、枯草菌胞子の胞子形成と発芽の両方において、それぞれDPAの取り込みと放出を促進することにより、重要な役割を果たしている(Gao, Barajas-Ornelas, et al, 2022; Setlow et al, 2017)。このチャネルを欠く胞子は不安定で自然発芽しやすく(Gao, Barajas-Ornelas, et al., 2022; Tovar-Rojo et al., 2002)、休眠胞子の安定化に重要な役割を果たしていることが示唆される。CとEBは複数のTMらせんを持つパラロガス膜タンパク質であり、Dはチオラーゼフォールドタンパク質と構造相同性を持つ球状の可溶性タンパク質である(Li et al.) 最近、GR依存的な胞子発芽の際、DはC-EB膜貫通複合体から放出され、DPAの放出を促進すると提唱されている(Gao, Barajas-Ornelas, et al., 2022)。こうして我々は、C、D、EBによって形成される高次複合体の4次構造を推測するために、このアプローチを用いた。複合体のサブユニット間の推定されるヘテロマー界面を同定するために、C-D、D-EB、C-EB間で共進化する残基対を検出した。(Gao, Barajas-Ornelas, et al., 2022)に記載されているように、C, D, EBの1:1:1複合体予測では、Dの推定ホモオリゴマー化界面やC-EB膜複合体のヘテロオリゴマー化界面にマップされるフリーコンタクトサイトがかなり見つかり(図5d)、複合体が不完全である可能性が示された。これらの共進化残基は、C-D-EBの2:2:2複合体のAF2-多量体予測におけるネイティブな分子間接触に対応している(図5e)。この高次複合体は、1:1:1複合体よりもipTMが改善され、AF2によって非常に正確に予測された。したがって、C-D-EBは「ヘテロ三量体の二量体」と会合してDPAチャネルを形成すると予測される(図6b)。Dの自己相互作用を仲介すると思われるネイティブなホモマー接触は、構造モデルにおいて2526Å2を埋める広範な界面にマップされる。各Cサブユニットはさらに、その中央のTMヘリックスを通して両方のEBサブユニットと接触し、またその逆も同様で、DPA輸送のための現在未知のチャネルを構成する可能性のある中央空洞を取り囲んでいる。我々のモデルは、この複合体のin vivo解析によって同定されたC-D-EB間の相互作用と一致しており(Gao, Barajas-Ornelas, et al., 2022)、DPAチャネルを構成する成分間のさらなる相互作用を予測している。
3.3.3 SpoVAAとSpoVABは相互作用し、4:2の化学量論的複合体を形成すると予測される
spoVAオペロンはC-D-EB(図5a)以外にも4つの遺伝子をコードしており、これらはDPAチャネルと機能的に関連している(Christie & Setlow, 2020)が、これらのタンパク質の機能や構造は不明である。そこで次に、これらの他のSpoVAタンパク質が、C-D-EBによって形成されるDPAチャネルと相互作用するのか、あるいは彼ら自身が特定の4元構造を持つサブコンプレックスを形成する可能性があるのかを予測した。予測された膜タンパク質SpoVAAとSpoVAB(それぞれAおよびB、図5f)は、以前(Gao, Barajas-Ornelas, et al., 2022)に仮説が立てられたように相互作用し(ipTM = 0.86)(図S7A)、したがってタンパク質複合体を形成する可能性があることだけがわかった。AF2-multimerは、残りのSpoVAタンパク質間のさらなる相互作用について確信に満ちた予測をしなかった(表S4)。したがって、spoVAオペロンは、A-BとC-D-EBによって形成される2つの別々のヘテロマー複合体をコードしているようである。興味深いことに、A-B間の共進化するネイティブな分子間コンタクトの最適な状態は、化学量論が均等な複合体よりもAF2-多量体予測の信頼性が向上した、化学量論が4:2の不均等な高次複合体においてのみ到達する(図5g、図S7a-d)。AとBのこの4:2複合体は擬4回環対称性を示し、各Aサブユニットの球状N末端ドメインとらせん状C末端ドメインは、複合体中の隣接する2つのAサブユニットに接触すると予測される(図6c)。さらに、BサブユニットはAの膜貫通らせんと十字型の孔のような構造に結合すると予測される(図6c)。したがって、我々のde novo構造予測と共進化解析アプローチは、ヘテロマー複合体予測における不均一な化学量論(Marsh et al.
4 結論
AF2は当初、タンパク質単量体のde novo構造予測用に設計され、トレーニングされてきた(Jumper et al., 2021)が、そのアルゴリズムはタンパク質複合体のモデリングも可能である(Evans et al., 2022)。そのため、AF2ベースの手法は、タンパク質-タンパク質界面における残基レベルの相互作用を同定するツールとして急速に普及している(Bryant, Pozzati, & Elofsson, 2022; Burke et al.) タンパク質複合体の化学量論が実験的にわかっている場合、AF2(-multimer)はタンパク質複合体の完全な四次構造を予測するために適用できる(Bryant, Pozzati, Zhu, et al., 2022; Evans et al., 2022)。しかし、AF2-multimerアルゴリズムが化学量論が未知のタンパク質の複合体を確実に予測できるかどうかは不明であった。我々の研究では、自明な化学量論でないタンパク質複合体のホモオリゴマーとヘテロオリゴマーを確実に予測することができたので、これは実際に可能である可能性を示唆している。これは、タンパク質複合体の分子間界面において、共進化するネイティブコンタクトの割合を追跡する共進化解析から得られた、モデル駆動型の解釈可能なスコアによって達成された。このスコアは、既存のAF2-多量体信頼性評価指標を補強するもので、複合体予測の化学量論的信頼性を記述するためのベンチマークは行われていない。多くの共進化残基対は、我々の構造予測におけるネイティブな分子内・分子間接触に対応し、我々のアプローチによって推定された化学量論的構造を持つ複合体は、通常、不完全な複合体よりもipTMスコアがわずかに向上した。
AF2-multimerの信頼性指標は、徐々に完全な部分複合体ほどわずかに向上することから、単なるノイズではなく「シグナル」であると考えられる。我々は、AF2-multimerアルゴリズムは、共進化する残基のネイティブな接触を暗黙のうちに最適化しているのではないかと推測している。このことは、一般にAF2が一貫して正確なde novo構造予測を行うためには、入力MSAに暗黙の共進化情報が必要であるという先行仮説と一致する(Jumper et al, 2021; Roney & Ovchinnikov, 2022)。このように、AF2-多量体信頼度が不完全なサブコンプレックスに対してわずかな変化しか示さない領域において、共進化に基づく解析は、より顕著で解釈可能なシグナルを提供する。インターフェイスの共進化解析は、不完全なサブコンプレックスを同定することによって化学量論が「過小評価」されるのを防ぐための解釈可能なスコアを提供し、AF2-マルチマーメトリクスは化学量論が「過大評価」されるのを防ぐことができる。
このことから、AF2-多量体信頼度スコアだけで、タンパク質複合体の化学量論的推定にある程度の予測力を持つ可能性がある。一般に、ipTMが最も高い複合体では、共進化するネイティブな分子間接触が最適であることがわかった。しかし、枯草菌GerAAの予測では、この傾向からわずかに逸脱していることが観察された。ホモ5量体はAF2-多量体の信頼性が最適であるが、共進化解析では、共進化残基の鎖間接触の割合がわずかに改善されたことから、ホモ6量体を示唆している。どちらのアプローチがより頑健であるかは、大規模なベンチマーク(下記参照)によって示される必要がある。この仮説を確認し、共進化に基づく手法がDeep-Learningベースのde novo構造予測とどのようにインターフェースするかを明らかにするために、この方向で将来研究を行う十分な機会があると期待している。
AF2-多量体構造モデルの分子間界面で共進化する残基ペアを可視化し、スコアリングすることで、例えば遊離オリゴマー化界面を検出することにより、複合体予測の解釈可能性が高まる(Anishchenko et al.) 我々は、化学量論が既知と未知の両方の複合体を用いて、自由接触が不完全なホモオリゴマーとヘテロオリゴマー複合体をピンポイントで特定することを示した。しかしながら、すべての複合体予測において、遊離接触部位がわずかに残っている。我々は、これらの共進化残基対の相当量は、AF2やAF2-multimerではサンプルされない、これらの複合体の代替コンフォーメーション(Anishchenko et al. 例えば、AF2は一般的にタンパク質のアポ型よりもホロ型を予測するように偏っており(Saldanõ et al.、2022)、BsAldの予測でも同様のパターンが観察された。 また、DがC-EBに結合した閉じたSpoVA DPAチャネルの予測では、自由な接触部位が多く観察され、開いた状態ではC-EBポアが別のコンフォメーションをとる可能性が示唆された。今後、タンパク質複合体のde novo構造予測の進歩により、このような別のコンフォメーションのサンプリングが容易になることが期待される。さらに、このような進歩により、タンパク質複合体の4次構造のより正確なde novo構造予測が容易になるかもしれない。
一般的に共進化解析は、共進化残基を確実に検出するために、かなりのMSAの深さと系統的多様性を必要とする(Avila-Herrera & Pollard, 2015; Hopf et al.) そのため、この手法は、相同配列がわずかしかないオーファンタンパク質や小さなタンパク質ファミリーには適用できない。細菌については、多くのゲノムが配列決定されており、そのゲノム構成は、未知の相互作用をスクリーニングするためのMSAペアリングを容易にする(Cong et al.) 対照的に、共進化解析は真核生物タンパク質ではより困難である。真核生物ゲノムの塩基配列は少なく、ゲノム構造はかなり複雑である可能性があるからである(Humphreys et al.、2021)。ディープラーニングに基づく鎖間接触予測アプローチ(Guo et al., 2022; Xie & Xu, 2022; Yan & Huang, 2021)は、利用可能な配列が少ないタンパク質複合体において、この制限を克服できる可能性がある。
我々のアプローチの現在の限界は、DCAによって同定された共進化残基のネイティブコンタクトの割合をスコア化するために、サブユニットの化学量論が異なる複合体の構造予測をAF2-multimerによって別々に行わなければならないことである。これは、多くの(サブ)複合体構成を構造モデル化する必要があるため計算コストが高く、また、現在の実装では、共進化するネイティブコンタクトのスコアリングに手作業が必要である。原理的には、これを自動化することで、タンパク質複合体の4次構造と化学量論のエンドツーエンドの予測を実現し、大規模なデータセットへの適用を容易にすることができるが、これは本研究の範囲外である。AF2-およびAF2-多量体を用いたタンパク質間相互作用のスクリーニングとモデル化は、計算コストのかかるde novo構造予測プロセスのため、既知の二項間相互作用に対するプレフィルターによってのみプロテオームに適用できる(Burke et al.) 対照的に、共進化解析は、高品質のMSAで多くの相同配列が利用可能であれば、プロテオームの大部分を容易にカバーすることができ、未知の相互作用をインシリコでスクリーニングすることができる(Cong et al.) 従って、我々のde novo四次構造予測戦略を大規模な検証データセットやプロテオームに拡張するには、(a)大規模な共進化解析のための高品質なMSAの慎重なアセンブリ、(b)検証された相互作用やその他の先験的知識によるタンパク質(サブ)複合体構造の予測値の厳格な事前フィルタリング、(c)予測された複合体上の共進化するネイティブな接触のスコアリングの完全自動化が必要である。
われわれの今回の研究が、生命科学の研究者たちが、未知の4次構造を持つ個々の細菌タンパク質複合体に対して、より小規模にわれわれの戦略を使用するきっかけになることを期待している。我々の戦略は、アクセス可能なオープンソースの2つの手法を組み合わせることに純粋に基づいたものである。AF2-multimerは、高性能計算システムやウェブベースの実装を用いて、すでに多くの生命科学研究者に広く使われている(Mirdita et al.) 我々の研究で使用されているDCAを用いた共進化解析は、デスクトップワークステーション上、または開発者が提供する一般にアクセス可能なウェブサーバー上で、統合されたPythonフレームワーク(Hopf et al., 2019)を用いて実施することができる(https://evcouplings.org/)。応用研究課題への我々の戦略の適用性を説明するために、枯草菌の膜関連胞子発芽タンパク質複合体の未知の四次構造を予測した。これらの解析に対応するソースファイルは、ソースデータに掲載されている(data availability statementを参照)。
GRのAサブユニットが自己相互作用して高次オリゴマーを形成しうるという我々の発見は、in vivoにおけるGRのクラスタリング(Griffiths et al. GerAAがホモオリゴマーを形成し、栄養ゲートイオンチャネルとして機能する可能性が、生化学的およびin vivoの新たな証拠から実際に示唆されている(Gao et al.) この研究の著者らは、AF2-多量体予測や限られた生化学的証拠に基づいて、GerAAがホモ-ペンタマーを形成することを示唆している。しかしながら、これは幾何学的な理由から、in vivoにおけるGRのクラスター形成と一致させることは困難である。我々の共進化解析とAF2-多量体構造予測との組み合わせから、GerAAのホモ6量体も同様に妥当であることが示され、大腸菌の化学受容体のようにGRが6角形の配列でクラスター形成する可能性がある(Sourjik & Armitage, 2010)。これらの可能性を区別し、胚芽体におけるGRのトポロジーを解明するためには、今後の実験的研究が必要である。
さらに、枯草菌のspoVAオペロンには、SpoVAA-BとSpoVAC-D-EBによって形成される、少なくとも2つのヘテロオリゴマータンパク質複合体がコードされている。我々の観察によると、これらのタンパク質が共同で1つのヘテロマー複合体を形成している可能性は低いが、胞子中のDPA輸送を促進するために機能的に連結しているようである(Gao, Barajas-Ornelas, et al., 2022)。C-D-EBによって形成されるDPAチャネルは「ヘテロ三量体の二量体」である可能性があり、このことは、チャネル内にさらなるタンパク質-タンパク質界面が存在し、その閉じた状態を安定化させ、ゲート機構に役割を果たしている可能性を示唆している(Gao, Barajas-Ornelas, et al.) この界面はDのホモ二量体X線結晶構造と構造的に類似しているが、精製されたDは溶液中では単量体であることが報告されている(Li et al.) DPAは胞子の高い耐湿熱性と安定性に関連していることから(Magge et al., 2008; Paidhungat et al., 2000; Setlow et al., 2017)、DPAを胞子内に保持し、休眠性と抵抗性を維持するためには、Dのこの界面が重要であると考えられる。
このように、今回のde novo四次構造予測は、部位特異的変異導入によるin vivoでのGRとSpoVA複合体の機能のさらなる特徴づけに役立つ可能性がある。この戦略は、何十年もの間、機構的な解析が不十分なままであった他の胞子発芽タンパク質にも拡張され(Christie & Setlow, 2020)、細菌の胞子発芽の分子機構の解明に役立つ可能性がある。
5 方法
5.1 AlphaFold2による構造予測
タンパク質モノマーと複合体の構造は、AlphaFold v2.2.0 (Evans et al., 2022; Jumper et al., 2021)を用いてモデル化した。モノマー構造は "monomer_ptm "パイプライン、すなわちAF2で予測し、複合体は "multimer "パイプライン、すなわちAF2-multimerで予測した。B.subtilisのクエリタンパク質配列はSubtiWiki (Pedreira et al., 2022)から取得し、クエリタンパク質の入力化学量論はタンパク質複合体予測のために手動で設定した。MSAは全遺伝子データベースセットを用いてクエリー配列上に構築した。その後、DeepMindが提供する5つのモデルパラメータセットそれぞれについて、テンプレートなしで3つの個別の構造予測を生成した。ただし、総アミノ酸数が2000を超える大規模な予測については、パラメータセットごとに単一の構造予測のみを行った。すべてのモデルは、AF2 v2.2.0に実装されているAmber力場で緩和された。すべての予測は、2つのNvidia A100 SXM4 GPU、36コアのIntel Xeon Platinum 8360Y CPU、および256GB RAMを使用する高性能計算ノードで、24時間の時間制限があった。構造予測は、単量体予測では全残基の平均pLDDT(Jumper et al., 2021)、複合体予測ではipTMとpTMの加重和(0.8×ipTM + 0.2×pTM)(Evans et al., 2022)でランク付けした。上位にランクされた構造予測のみを詳細に評価した。
5.2 モノマーおよびペア多重配列アラインメントの構築
共進化残基は、以前に記述したように、EVcouplings v0.1.1 (Hopf et al., 2019)を用いてplmDCAで検出した(Artzi et al., 2021; Gao, Barajas-Ornelas, et al., 2022)。すべてのクエリタンパク質配列について、2022年6月にダウンロードしたUniRef100(Suzek et al.、2015)に対して5回の反復を行い、jackhmmer(Johnson et al.、2010)を用いてモノマーMSAを構築した。異なる深さのMSAを、異なるbitscore包含閾値(クエリの配列長の0.1、0.3、0.5、0.7、0.8倍)で、少なくとも70%の非ギャップ文字のカラムカバレッジで生成した。クエリー配列の少なくとも70%をカバーしない整列配列は破棄された。ヒットは90%配列同一性カットオフでクラスタ化され、冗長性を減らし、サンプリングされた配列の系統的多様性を広げるために、まとめてダウンウェイトされた。モノマーMSAのヒットは、UniProt IDに基づいてアノテーションされたクエリと同様のPfam IDおよびInterPro IDがあるかどうかをチェックした。クエリーとしての胞子発芽タンパク質については、既知の胞子形成細菌に属さない細菌由来の機能的に無関係なリモートホモログが含まれないように、ヒットがBacillota門の種由来であることを確認した(Galperin et al. そして、これらの基準を満たし、最も高い配列カバー率と深さ(すなわちNeff/L)を持つモノマーMSAを選択した(表S1)。2つの異なるタンパク質間で共進化している残基を検出するために、クエリータンパク質配列の両方について、2つのモノマーMSAの種ごとの相互ベストヒットをペアにした(Green et al., 2021)。クエリー配列と95%以上の配列同一性を持つ相互ベストヒットは、このペアリング手順から除外した。ペアリングされたMSAは最小カラムカバレッジ閾値50%でフィルターされ、ペアリングされた配列は90%の同一性カットオフでクラスタ化された(表S2)。
5.3 擬似尤度最大化直接結合解析による共進化残基の検出
擬似尤度最大化DCA(Hopf et al., 2017, 2019)を用いて最大エントロピーモデルパラメータ(Balakrishnan et al., 2011; Marks et al., 2011)を推論し、100反復、正則化パラメータ(λhおよびλJ)を0.01に設定して、MSAにおける残基-残基共進化を検出した。クエリの残基のすべての相互作用の可能性のあるペアの間の結合強度は、以前に記載されたように、補正ノルムスコア(cn-スコア)として表された(Ekebergら、2013)。cn-スコア分布は、最尤推定により、低スコアのスキュー正規分布成分と高スコアの対数正規分布尾部を含む混合モデルでフィッティングした。クエリー配列で少なくとも6アミノ酸離れている残基対は、関連するcn-スコアがこのフィットされた混合モデルの対数正規成分に属する事後確率が0.9を超える場合のみ、有意なヒットとした(Hopf et al., 2014; Toth-Petroczy et al.) すべての有意なヒットをそれぞれのcn-スコアで降順にランク付けし、上位の共進化残基ペアを同定した。
5.4 Alphafold2構造予測における共進化残基ペアのマッピング
共進化残基は、EVcouplings v0.1.1 (Hopf et al., 2019)の構造解析ツールを用いて、AF2およびAF2-マルチマー構造予測にマッピングした。i)複合体の各サブユニットにおける鎖内距離、ii)サブユニットの全コピー間のホモマー分子間距離、iii)ヘテロマー複合体における2つの異なるサブユニットの全ユニークな組み合わせ間のヘテロマー分子間距離を区別した。距離マップは、AF2およびAF2-多量体予測におけるそれぞれのタンパク質サブユニット間の重原子間の全対全対最小残基間距離を計算することによって計算された。それぞれの距離マップからの最小残基間距離に基づいて、共進化残基ペアを分子内相互作用ペアまたは分子間相互作用ペアのいずれかに割り当てた。共進化残基対は、計算された最小距離が5Å未満であれば共進化ネイティブ接触と定義し(Anishchenko et al., 2017; Méndez et al., 2003)、8Å以上離れていれば非ネイティブ、フリー接触部位と定義した(Artzi et al., 2021; Hopf et al., 2014; Morcos et al., 2011)。
5.5 多量体構造の解析と可視化
AF2およびAF2-多量体構造予測は、UCSF ChimeraX 1.5 (Pettersen et al., 2021)で可視化および解析した。共進化残基対は、側鎖を省略し、共進化残基対のCα原子間に擬似結合を描くことにより、複合体上にプロットされた。複合体中の接触表面は、プローブ半径1.4Åを用いて、鎖対のすべての対の溶媒アクセス可能な表面を計算することにより同定した。界面におけるサブユニット間の接触表面は300Å2以上埋まる必要があり、これらの界面内の15Å2以上埋まった残基を界面の接触表面残基として分類した。AF2で予測された構造と実験的に決定された構造を比較するために、US-align (Zhang et al., 2022)を用いてTM-score (Zhang & Skolnick, 2004)とCα-RMSDを測定した。pTM、ipTM、pLDDT、PAEなどのAF2およびAF2-multimerメトリックスと、AF2予測における共進化残基の接触スコアリング結果は、matplotlib 3.6.2を用いて可視化した。
著者貢献
マックス・キリアン 概念化、調査、原案執筆、校閲・編集、可視化、ソフトウェア、形式的解析。イルカ・B・ビショフス: 概念化、監修、プロジェクト管理、執筆-原案、執筆-校閲・編集。
謝辞
計算はドイツ、ガルヒングのMax Planck Computing and Data Facilityの高性能計算システムRavenで行った。本研究はマックス・プランク協会(I.B.B.)の資金援助により行われた。本研究は、M.K.がドイツ、ハイデルベルク大学薬学・分子バイオテクノロジー研究所(IPMB)の理学修士号(MSc)の要件の一部を満たすために実施したものである。Projekt DEALによるオープンアクセス助成を受けた。
資金提供情報
資金提供者は、研究デザイン、データ収集と分析、出版決定、原稿作成には関与していない。
利益相反声明
著者らは、競合する利益はないと宣言している。
倫理声明
本論文には、人体または動物を用いた研究は含まれていない。
公開研究
サポート情報
参考文献
PDFダウンロード
戻る
その他のリンク
ワイリーオンラインライブラリーについて
プライバシーポリシー
利用規約
クッキーについて
クッキーの管理
アクセシビリティ
ワイリーリサーチDE&Iステートメントと出版ポリシー
発展途上国へのアクセス
ヘルプ&サポート
お問い合わせ
トレーニングとサポート
DMCAと著作権侵害の報告
チャンス
購読エージェント
広告主・企業パートナー
ワイリーとつながる
ワイリーネットワーク
ワイリープレスルーム
著作権 © 1999-2023 John Wiley & Sons, Inc. すべての著作権はワイリーに帰属します。