
初心者の菌叢解析 Qiime2で解析(7)fastqファイルの取り込み(変換)

本日から実際に解析を進めていきます。
最初の目標は、菌種の組成を示すグラフを作るところを考えています。
その後に、多様性解析やPICRSt2を動かしていこうと思います。
今回はfastqファイルの取り込み(アーティファクト[.qza]への変換)を行います。
流れとしては、manifestファイルを元に、fastqファイルの取込(変換)を行います。その後、クオリティ等を確認したいと思います。
1.はじめに
Qiime2の動かし方ですが、基本的にまずは「qiime」というコマンドを入れます。その後ろに、様々なコマンドを追加し、実際の解析やプラグインを動かしていきます。
そして、解析を進めると2種類の拡張子でファイルが生成されます。また、Qiime2で生成されたファイルは「アーティファクト」と呼ばれます。
1-1.[.qza]ファイル
Qiime2で生成されるメインのファイル。
機械語で書かれている為、内容はよく分からない。生成されたqzaファイルを使用して次のqzaファイルを生成し、解析を進めていく。
1-2.[.qzv]ファイル
qzaファイルを可視化したファイル。
いわゆる結果のデータにあたり、以下の可視化サイト(Qiime2 View)にドラッグ&ドロップすると内容が見られる。出てきたらとにかく見てみると楽しい。
2.ターミナルの起動とディレクトリ移動
ターミナルを起動し、「Qiime2_test」に移動します。
ここで、「Qiime2_test」というディレクトリの中に移動したことになっており、ターミナル上の現在地(PWD)が「Qiime2_test」になります。
cd /Users/ユーザー名/Desktop/Qiime2_test 以下のコマンドを入れれば現在地が表示されると思います。
pwd次にQiime2環境をアクティベートします。
conda activate qiime2-2021.23.fastqファイルの取り込み
Qiime2にfastqファイルを取り込みます。以下のコマンドがうまく働けば、緑色の字が出てきてフォルダ内に「paired-end-demux.qza」というファイルが生成されます。また、こちらのコマンドはペアエンドの配列のみに対応しています。
qiime tools import \
--type 'SampleData[PairedEndSequencesWithQuality]' \
--input-path manifest.csv \
--output-path paired-end-demux.qza \
--input-format PairedEndFastqManifestPhred33エラーが出た場合は以下に問題があると思います。
1.コマンドが間違っている。
2.fastqファイルが間違っている。(形式を確認)
3.manifestファイルが間違っている。
基本的にはエラーコードにその失敗内容が書いてあります。
ですので、そちらを読んでなんとなくエラー内容を確認する必要があります。もしよろしければ、エラーコードが出たらコメントに書き込んでください。出来れば解決方法も書いていただけますと助かります。
余裕があったら記事内容に追記させていただきます。
一番多いエラーはmanifestファイルに問題があることだと思います。
その時は以下の記事のエラーコードに関する部分を確認してみてください。
「paired-end-demux.qza」が生成されたら以下のコマンドを入れます。「demux.qzv」というファイルが出てくると思います。
qiime demux summarize \
--i-data paired-end-demux.qza \
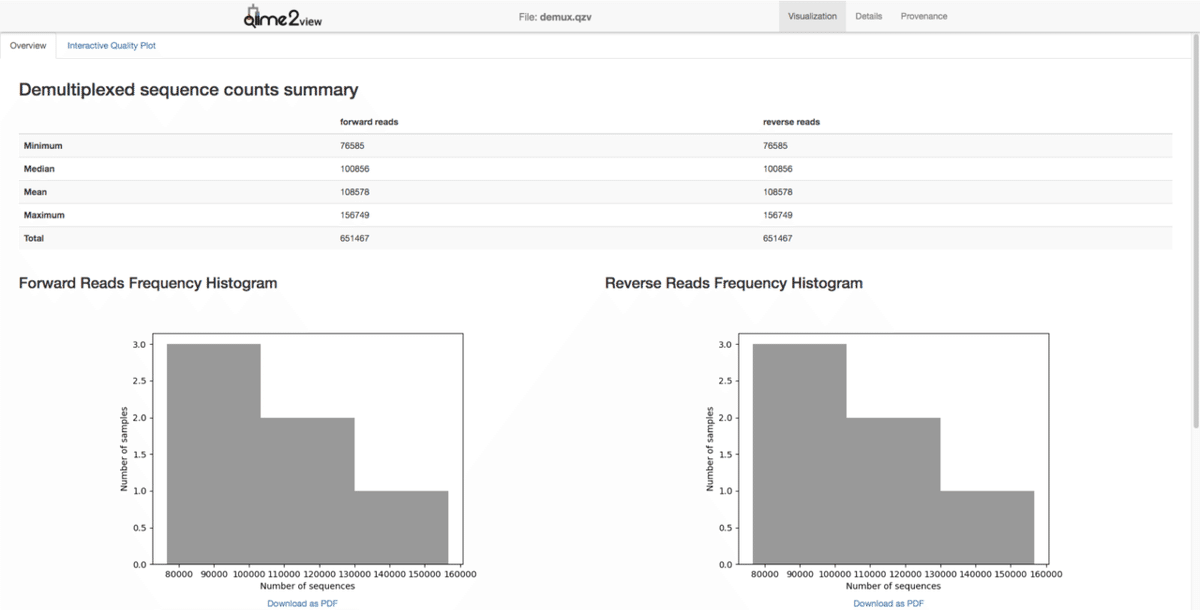
--o-visualization demux.qzv「demux.qzv」というファイルは可視化ファイルなので、早速Qiime2 Viewで確認します。
Qiime2 Viewに「demux.qzv」をドラッグ&ドロップします。

qzaファイルに変換した内容について確認できます。
上のタブを切り替えて「Interactive Quality Plot」をクリックすればfastqファイルのクオリティを確認できます。左がforward配列で右がreverse配列です。クオリティが配列が進めば進むほど低下していくことが確認できます。
データは箱ひげ図ですので、クリックしたり拡大したりして詳細が見られます。また、下にはデータをまとめが表がありますので、各baseのクオリティを確認できます。

次回はこのクオリティを元にDADA2を動かしていきます。