
初心者の菌叢解析Qiime2 classifierを自作する 改訂版

以前もQiime2のclasifier.qza作成方法について投稿しております。
しかし時間が経ち、ダウンロードができない等の不都合が出てきましたので、改訂版を投稿いたします。
Classifier作成には意外と時間がかかりました。
出来合の物で問題がなければ本投稿の「5-2.full lengthで良ければダウンロードできる」までお進みください。
1.データベースについて
使用するデータベースは基本的には「GreenGenes」を使用したものが多いです。しかし、GreenGenesがなかなか更新されないデータベースであるため、ここでは「Silva」を用いたclasifier.qzaの作成を進めてみます。
2.Qiime2 docsから16S rRNA配列のqzaファイルをダウンロード
まずは16S rRNA配列のデータをダウンロードします。
基本的には配列のデータなので、fastaファイルなのですが、Qiime2 docに既にqzaファイルに変換したものがありました。
具体的には、「Silva」の最新データがありまして、そちらを使用します。
さらに、Taxonomyのqzaファイルもありましたので、同時にダウンロードします。また、Qiime2 docsは最新のバージョンを確認してください。
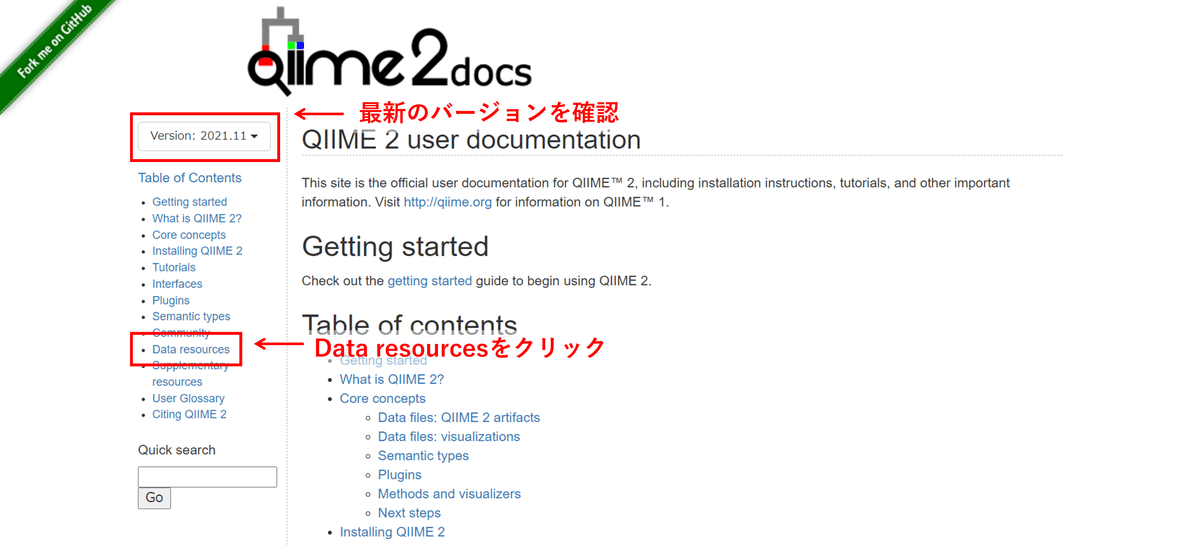
上の画像の通り、Qiime2 docsのバージョンを確認後、「Data resources」をクリックします。
次に下へスクロールしていき、「Silva (16S/18S rRNA)」の項目から以下の2つのファイルをダウンロードします。
1.Silva 138 SSURef NR99 full-length sequences
2.Silva 138 SSURef NR99 full-length taxonomy

ダウンロードしたら、ファイルを目的のフォルダに移動しておきます。
私の場合は以前の投稿で作成した「Qiime2_test」フォルダになります。
3.16S rRNA配列上から今回の解析に必要な部分を取り出し、classifier.qzaファイルを作成
macの場合はターミナル、Windowsの場合はUbuntuを起動し目的のフォルダに移動します。その後、以下のコマンドでQiime2をアクティベートします。
cd /Users/ユーザー名/Desktop/Qiime2_test
conda activate qiime2-2021.2それぞれのQiime2インストールについては過去の投稿をご確認ください。
Qiime2を起動後、ダウンロードしたqzaファイルからClassifierを作成していきますが、いくつか注意点があります。
Classifierを自作する際の一番のポイント
サンプル(腸管内容物や糞便等)からTotal DNAを抽出した後に、16S rRNA配列の一部を1'st PCRにて増幅していると思います。
その時に使用したプライマーの配列を次のコマンドで入力します。
ただし、入力するのは「16S rRNA配列に結合するプライマーの配列」のみになります(重要)。
1'st PCRで用いるプライマーには2'nd PCRでIndex配列を付加するための付加配列が含まれている場合が多いと思います。
そのため、16S rRNA配列上から今回の解析に必要な部分を取り出し、classifier.qzaファイルを作成する場合はこの付加配列は必要ありません。
16S rRNA配列に結合するプライマーの配列をデータシートで確認するか、プライマー配列を販売しているメーカーに問い合わせれば分かると思います。
このことを踏まえて以下のコマンドを実行します(結構時間かかります)。
イルミナのV3-V4領域で菌叢解析をする場合、以下のコマンドで使用している配列になると思います。プライマー配列の3’側が以下の配列と一致しているはずですので、確認してみてください。
qiime feature-classifier extract-reads \
--i-sequences silva-138-99-seqs.qza \
--p-f-primer 「Forward primerの配列を入れる」例: CCTACGGGNGGCWGCAG \
--p-r-primer 「Reverse primerの配列を入れる」例: GACTACHVGGGTATCTAATCC \
--p-max-length 「ここには1'st PCR産物のサイズを入れる」 例: 600 \
--o-reads silva-138-99-ref-seqs.qza「--p-f-primer」の後にはForward primerの16S rRNA配列に対する結合配列を入力し(大文字)、「--p-r-primer」の後にはReverse primerの結合配列を入力します。
「--p-max-length」は入力した数字以上はカットするためのコマンドなので、1'st PCRのPCR産物のサイズ+αを入力します(私が多用するV3-4領域であれば、600 bpくらい)。
うまくいけば、「silva-138-99-ref-seqs.qza」が生成されます。
※この段階でエラーが出てしまった場合はコメント欄にも解説がありますので、参考にしてみてください。
また、「killed」と出た場合はPCのメモリ不足の可能性があります。
違うPCでclassifierの作成を行うか、出来合のファイルをダウンロードする必要があるかもしれません。
最後に以下のコマンドを実行し、終了(こちらも結構時間かかります)。
qiime feature-classifier fit-classifier-naive-bayes \
--i-reference-reads silva-138-99-ref-seqs.qza \
--i-reference-taxonomy silva-138-99-tax.qza \
--o-classifier silva-138-99-classifier.qza「silva-138-99-classifier.qza」ファイルが生成されれば終了なので、
このファイルを解析で使用する目的フォルダにコピペする。
4.taxa-bar-plotsの作成
作成したClassifierを用いてtaxa-bar-plotsを作成しました。一応比較として、以前作成した「Greengenes 13_8」でのClassifierでの解析も行ってみます。
taxa-bar-plotsの作成は以前の投稿をご確認ください。
「gg-13-8-99-515-806-nb-classifier.qza」の部分を「silva-138-99-classifier.qza」に変更して実施しています。
結果は以下の画像のようになりました。

左が今回作成したClassifierで解析した結果で、右がGreengenes 13_8をもとに作成したClassifierでの解析結果になります。
見て分かるように、Silvaで作成した左図の結果では分類がより細かくできていることが分かります。Level 2の段階でこの差が出ているので、かなりの違いだと思います。
また、左の赤矢印で示す「Marinimicrobia」は以前(右図)は「SAR406」というように表記されており、データベースの更新状況を感じます。
さらに、右図の矢印の分類は分類不可能であった配列ですが、左のSilvaでは格段に減少していることが分かります。
データベースの更新だけでなく、左図はV3-V4領域で最適化してあり、右図はV4のみのClassifierになっている為、このような差が生まれた可能性もあります。
5.Classifier自作のデメリット
今回作成してみていくつか感じたことがありましたので、デメリットとして記載します。
5-1.データ量が大きすぎる
データベースが更新されている為、データ量が「Greengenes 13_8」と比較し、大きくなっています。
そのためか、Classifier作成に非常に時間がかかりました。一度作成してしまえば当分は使える為、そんなには問題にならないかもしれませんが、私のスペックの低いWindowsの方では作成できませんでした。
※PCのメモリが足りないと、「killed」と出てしまうようです。
5-2.full lengthやV4領域に特化したもので良ければダウンロードできる
今回はV3-V4領域に最適化してClassifierを作成していますが、full lengthやV4領域のみで良ければQiime2 docsからダウンロードできます。
「Data resources」の中の「Silva 138 99% OTUs full-length sequences」もしくは下の「Silva 138 99% OTUs from 515F/806R region of sequences」を使用すれば解析可能だと思います。
データはそのうち更新されていくと思いますので、適宜確認してください。
一応その下にGreengenes 13_8のClassifierもあります。

6.classifierに関するエラーコード
これまでにであったエラーコードを記載しておきます。
taxonomyの割り振りをする作業をQiime2で今後行っていくのですが、
以前以下のようなエラーコードがでました。
"The scikit-learn version (0.20.2) used to generate this artifact does not match the current version of scikit-learn installed (0.23.1). Please retrain your classifier for your current deployment to prevent data-corruption errors."
Googleに尋ねたところ、Qiime2 forumで似たようなエラーコードの投稿がありました。それによると、Qiime2のバージョンが同じclassifier.qzaをダウンロードするか、自分でclassifier.qzaを作成してくださいとのことでした。
今回の投稿のように自作できれば問題は解決できると思います。
また、同じバージョンのQiime2 docsを開いてclassifierをダウンロードすれば大丈夫だと思います。
入力するプライマー配列に問題があると以下のエラーが出るようです。
Plugin error from feature-classifier:
No matches found
See above for debug info.
このようなエラーが出てしまう場合の対処方法は本投稿のコメント欄でも少し解説していますので、参考にしてみてください。
今回は以上になります。
見ていただきありがとうございました。