Rを使って多変量データのプロットと統計解析をする

はじめに
こんなときありませんか・・・?
様々な調査地で記録した生物群集の特徴を抽出し、2次元のプロットに要約したい
微生物相の構成に影響する要因を調べたい
脂肪酸組成の特徴からある動物の餌を推定したい
そこで今回は、多変量解析のためのRコードを紹介します。
ここではRコードの解説が中心になります。統計学に基づいた解説が必要な方は、こちらの論文をご参照ください。
生物群集解析のための類似度とその応用 : Rを使った類似度の算出、グラフ化、検定
土居 秀幸, 岡村 寛
https://doi.org/10.18960/seitai.61.1_3
準備する
次のようなシナリオを考えます。
①地域ごとにどのような生物が生息しているか調査し、生物相が似ている地域を見つける
②生物相の類似度に温度などの環境条件が関係しているかを調べる
データファイル
今回は地域ごとの生物相データを使って説明していきます。
以下のサイトでアップロードされているデータセットを使います。
Rによる群集組成の解析
データセットを2種類用意します。
データセットその1:地点ごとに生物群集をまとめたデータ
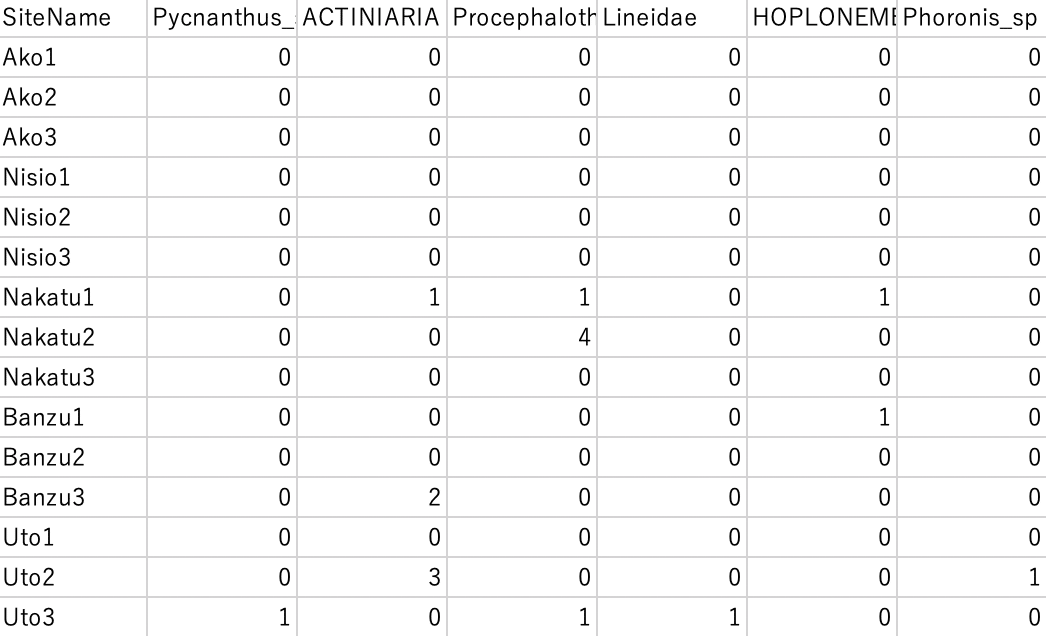
基本的な構成は
1列目:地点のID
2列目以降:生物種とその数
です。
※注意
地点IDは重複しないように付与しましょう
"spcdat.csv" と名前を付けて保存します。
データセットその2:各地点の環境条件をまとめたデータ
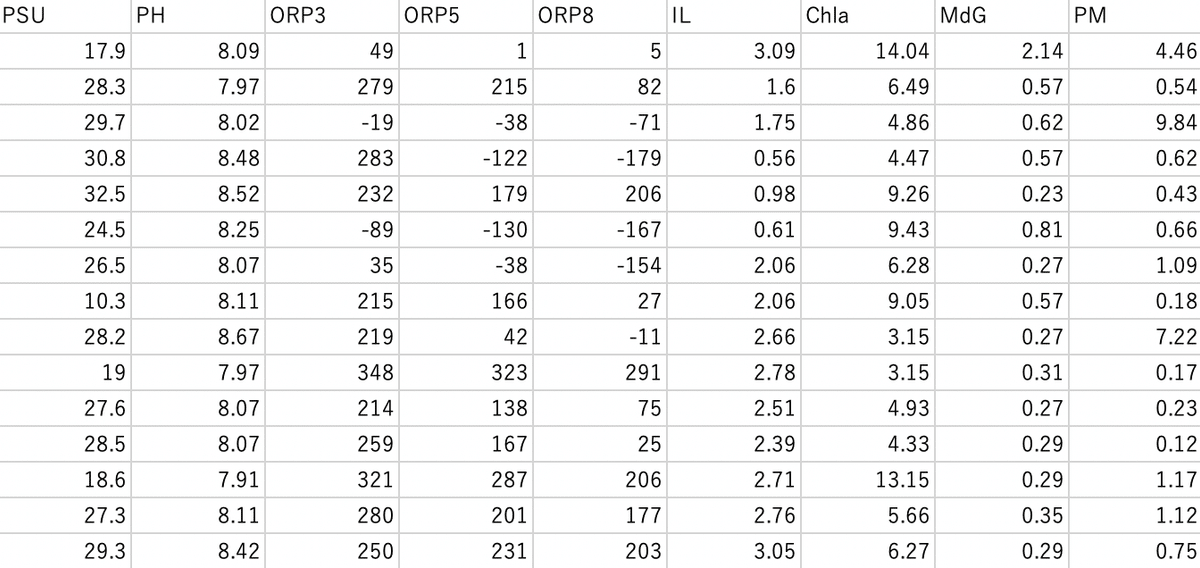
生物相を決めうる要因として、クロロフィル濃度やpHなどの環境条件に着目します。
※"spcdat.csv"の1列目で指定した地点IDに対応するように環境データを入力しましょう
"envdat.csv" と名前を付けて保存します。
vegan package を入手する
多変量解析では、vegan パッケージをフル活用します。
veganは群集生態学のデータ解析ために作られたRパッケージです。
vegan パッケージをインストールしていない場合は、以下のコードを実行してください。
install.packages("vegan")やってみよう
類似度の計算
生物相が地点間で「似ているか or 似ていないか」は、類似度指数を計算することで評価します。
これまで、いくつかのタイプの類似度指数が提案され、実際に生態学分野の研究で使われてきました。たとえば、Bray-Curtis index, Morishita index, Jaccard indexなどがあります。
今回は "Bray-Curtis index" を使って、類似度を計算します。
※注意
厳密には、Bray-Curtis indexでは非類似度を計算しています。
library(vegan)
fauna <- read.csv("spcdat.csv",header=T,row.names=1)
bcdist <- vegdist(fauna,"bray")
bcdist解説
#1 library(vegan) で、ライブラリーを呼び出します。
#2 read.csv() で、データファイルを読み込みます。
読み込んだデータファイルは、ひとまず "fauna" という「箱」に格納しておきます。
spcdat.csv データでは、1行目に種名、1列目に地点IDが記載されていますが、これらは類似度行列の計算では不要な情報です。
そのため、read.csv() で、header=T と row.names=1 を指定しました。
header では列名があるか無いかを指定します。
row.names ではIDが格納されている行番を指定します。今回のように、1行目が種名の場合は、1を入力しておきます。
#3 vegdist() で類似度を計算します。
この関数は、vegdist(データセット、指数のタイプ) のように使います。
指数のタイプには、以下のようなものがあります。
"bray" -> Bray-Curtis 指数
"morishita" -> Morishita 指数
"jaccard" -> Jaccard 指数
ここでは、Bray-Curtis 指数に基づいて類似度を算出しました。結果を "bcdist" という箱に格納しておきます。
#4 bcdist を実行すると地点間の類似度 (距離行列) が出力されます。
2次元プロットを作成する
先ほど計算した類似度を2次元にプロットしてみましょう。
多変量のデータを次元圧縮して図に落とすための手法はいくつかありますが、
ここでは群集生態学でよく使われる 非計量 (non-metric)-多次元尺度法 (multi-dimensional scaling) を使います。
つまり、nMDSをします。
bcmds <- metaMDS(bcdist, k = 2)
bcmds$stress
ordiplot(bcmds,type="n")
points(bcmds)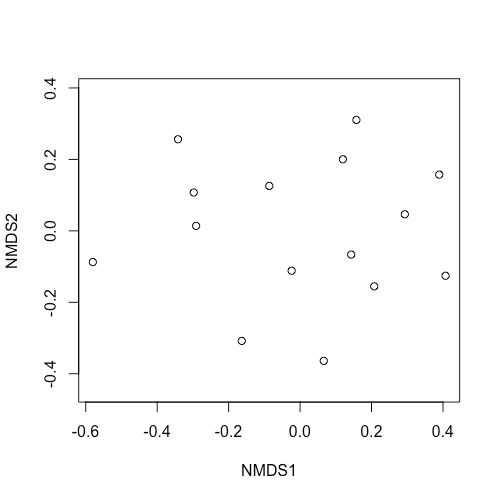
解説
#5 metaMDS() で類似度データをk次元に圧縮します。
あらかじめ計算しておいた類似度データ "bcdist" を metaMDS() 関数に入力しましたが、
metaMDS(fauna, dist = "bray", k = 2) としてもOKです。
*Note*
veganパッケージで用意されている metaMDS 関数を使ってnMDSをおこないましたが、
MASSパッケージの isoMDS 関数でもnMDSができます。
isoMDS(bcdist, k = 2) で計算できます。
#6 bcmds$stress でストレス値を確認しましょう。
ストレス値を(超)簡単に説明すると、類似度データをnMDSによってk次元に落とした際の「あてはまりの良さ」を表しています。
ストレス値が0.2より大きいと、あてはまりが悪いと判断されます。
#7 ordiplot() で "キャンバス"を用意します。
type = "n" とすることで、プロットは出力されず、軸の範囲が維持された状態で「白紙」ができます。
#8 points() で類似度データをプロットします。
基本的なプロットの解説はここまでです。
プロット色の変更など、図のカスタマイズ方法については記事の後半で紹介します。
データ解析をする
さて、出力されたnMDSの2次元プロットを眺めていると、いくつか疑問がわいてきます。
地点間の「似ているor似ていない」を決める生物種はなんだろう
生物相を決める要因はなんだろう
それでは、Rで解析してみましょう
ベクター解析
地点間の「似ているor似ていない」を決める生物種はなんだろう
これをベクター解析で調べてみましょう。
envNMDS <- envfit(bcmds, fauna)
ordiplot(bcmds,type="n")
points(bcmds)
plot(envNMDS, col="black", p.max=0.01)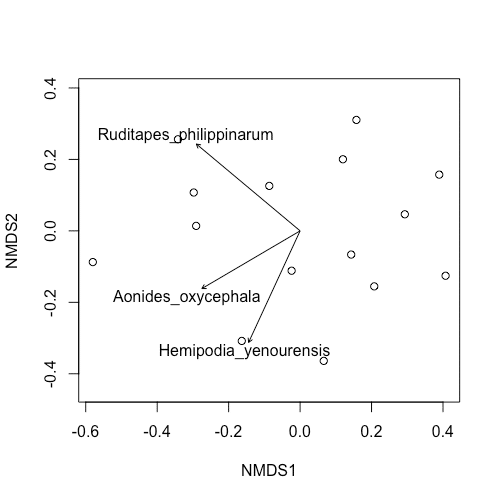
解説
#9 envfit() で nMDS で得られた配置と、ある要因の相関を調べることができます。
関数のルールは、envfit(類似度データ, 要因データ) です。
ここでは、類似度に関係する生物種を調べたいので、faunaデータセットを選択しました。
計算結果をenvNMDSという箱に格納しておきます。
*Note*
加えて、試行回数を設定することができます。デフォルトでは999回ですが、足りないという方は permu = で適当な値を指定してください。
例)envfit(bcmds, fauna, permu = 9999)
ベクターをプロットをしてみましょう
#10 ordiplot() で "キャンバス"を用意します。 (#7と同じ)
#11 points() でデータをプロットします。 (#8と同じ)
#12 plot(envNMDS, col="black", p.max=0.01) で先に計算した、各要素の座標をベクターとして出力します。
col = でベクターの色を指定します。
また、p.max = で p値をもとに出力するベクターを決めることができます。
要素を全て出力すると可読性が下がるため、有意に相関する要素だけ出力しました。
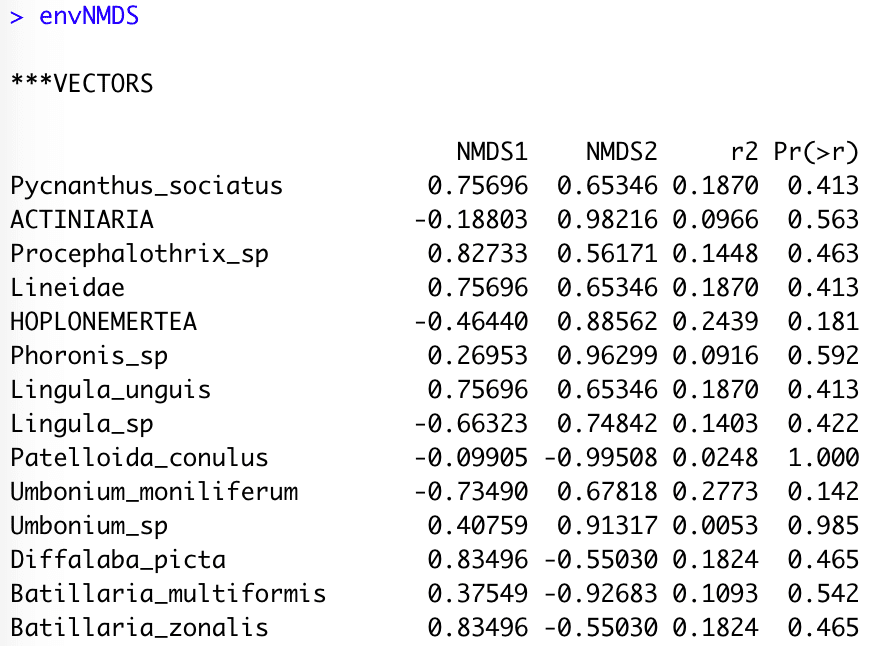
なお、各要素の座標、R^2、p値はenvNMDSを実行すれば確認できます。
生物相を決める要因はなんだろう
これも同様の手順で調べることができます。
env <- read.csv("envdat.csv", header = T)
envNMDS <-envfit(bcmds, env)
ordiplot(bcmds,type="n")
points(bcmds)
plot(envNMDS, col="black")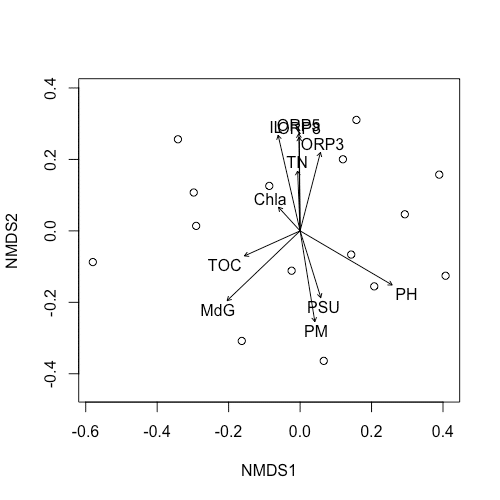
解説
#13 read.csv() で各地点の環境条件をまとめたデータ "envdat.csv"を読み込みます。(おまたせしました)
1行目に各要因の名前が入っているので、header = T を指定しておきます。
#14 envfit() で nMDS で得られた配置と、ある要因の相関を調べることができます。 (#9と同じ)
#15 ordiplot() で "キャンバス"を用意します。 (#7,10と同じ)
#16 points() でデータをプロットします。 (#8,11と同じ)
#17 plot(envNMDS, col="black") で各要素の座標をベクターとして出力します。(#12と同じ)
ただし、今回は p.max = を指定していません。
統計解析
さて、ここまでベクター解析の方法を紹介してきました。
しかし、残念なことに、nMDSで得られた「似ているor似ていない」を示す結果には統計的なパワーがありません。
そこで、類似度解析に役立つ統計解析を紹介します。
PERMANOVA
PERMANOVA (PERmutational Multivariate ANalysis Of VAriance) では、距離行列に対してノンパラメトリック (多変量) 分散分析を行うことができます。
*Note*
類似の解析手法としてANOSIMがありますが、これは諸事情により使用が推奨されていません。
それでは、次のようなシナリオを考えてみます。
faunaデータは、春と秋の2シーズンにわたって採取されたサンプルで、時期によって生物群集が異なるかを調べたいとします。
説明の便宜上、"fauna.csv" のデータを次の2グループに分けます。
→ Spring, Autumn
season <- c("Spring","Spring","Autumn",
"Spring","Autumn","Autumn",
"Spring","Spring","Autumn",
"Spring","Autumn","Autumn",
"Spring","Spring","Autumn")
adonis(fauna ~ season, method="bray")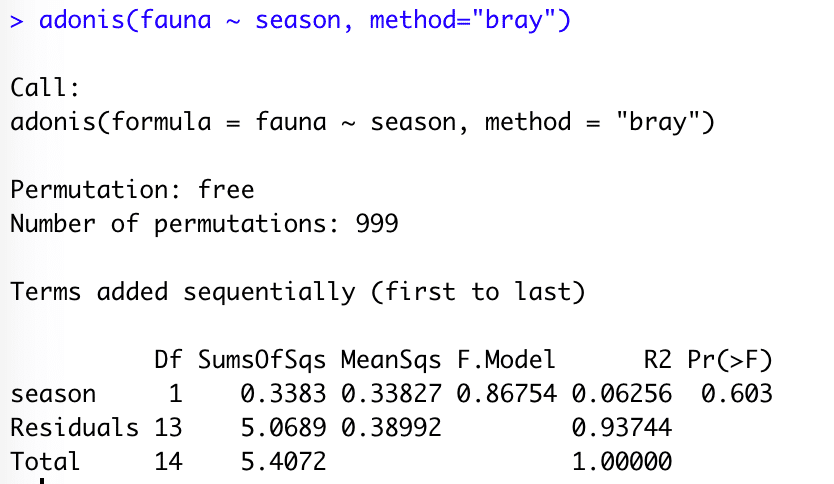
解説
#18-22 Ako1~Uto3のデータ列に時期の情報を付与するために、"Spring"または"Autumn"が格納されたデータリストを作りました。
#23 adonis() でPERMANOVAを実行します。
関数のルールは、adonis(群集データ, 要因データ) です。
関数に要因データを2つ以上組み込みたい場合は adonis(群集データ, 要因データ1 + 要因データ2) としましょう。
今回のように2グループ ["Spring"または"Autumn"] 間で比較する場合はadonis() でOKですが、
3グループ以上で比較する場合は pairwisePERMANOVAがおすすめです。
pairwisePERMANOVA
それでは、次のようなシナリオを考えてみます。
faunaデータは、5水域で採取されたサンプルであり、水域によって生物群集が異なるかを調べたいとします。
説明の便宜上、"fauna.csv" のデータを次の5グループに分けます。
→ Ako, Nisio, Nakatu, Banzu, Uto
site <- c("Ako","Ako","Ako",
"Nisio","Nisio","Nisio",
"Nakatu","Nakatu","Nakatu",
"Banzu","Banzu","Banzu",
"Uto","Uto","Uto")
library(pairwiseAdonis)
pairwise.adonis(fauna, site)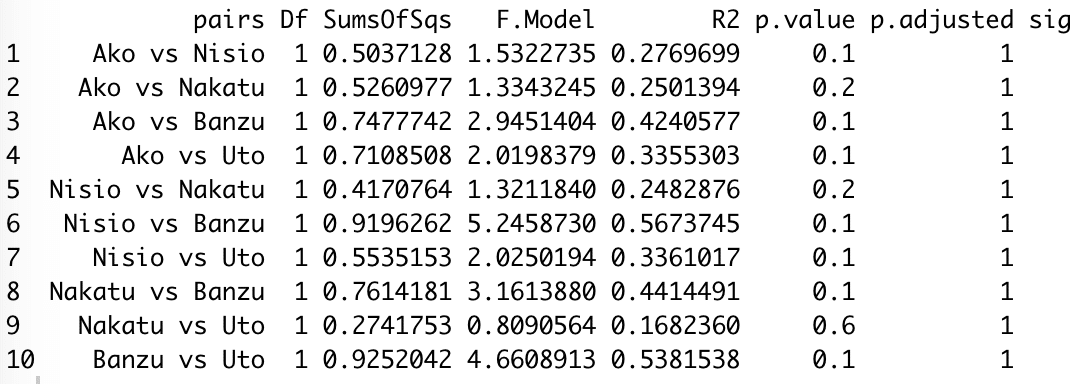
解説
#24-28 Ako1~Uto3のデータ列を地点5地点に分けるために、グループIDが格納されたデータリストを作りました。
#29 library() でライブラリーを呼び出します。
pairwiseAdonis をインストールしていない場合は、install.packages("pairwiseAdonis") を実行しましょう。
(github からのダウンロードなので、環境によってはこのコードではうまくいかないかも・・・お困りの方はコメントください)
※ pairwiseAdonis の github サイトはこちらhttps://github.com/pmartinezarbizu/pairwiseAdonis
#30 pairwise.adonis() でpairwisePERMANOVAを実行します。
関数のルールは、pairwise.adonis(群集データ, グループデータ) です。
2種類のp値が出力されますが、p.adjusted を使用することを推奨します。
Bonferroni 補正によって修正されています。
*Note*
今回使用したサンプルデータで実行すると、赤字でエラーメッセージが大量に生成されるかと思います。おそらく各グループのサンプルサイズが少ないためだと思われます。
SIMPER
ここで再び次のシナリオを考えます。
地点間の「似ているor似ていない」を決める生物種はなんだろう
それでは、SIMPER (Similarity Percentage) で、類似度に影響したカテゴリーの寄与度を調べてみましょう。
simper(fauna, site, permutations = 999)
sim <- simper(fauna, site, permutations = 999)
sim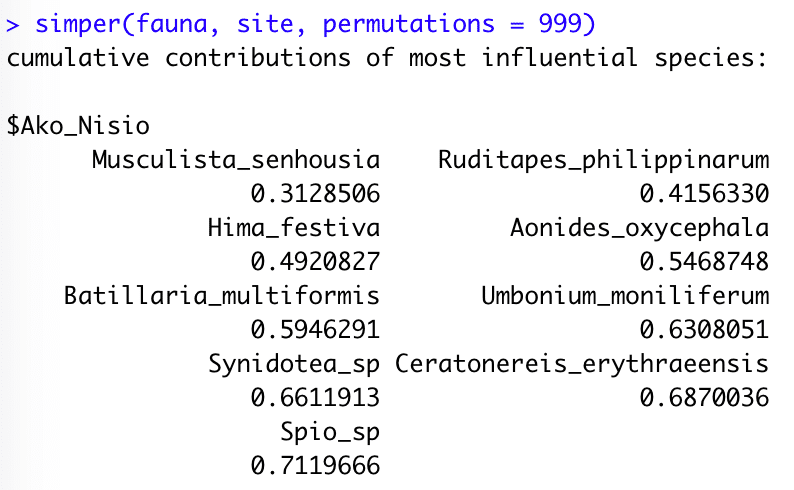
解説
#31 simper() で SIMPER を実行します。
関数のルールは、simper(群集データ, グループデータ) です。
群集データのカテゴリー(生物種)ごとに、グループ間の(非)類似度への寄与度が計算されます。
出力結果では、寄与度の大きいカテゴリーから順に並べられています。
今回のデータセットでは、Musculista_senhousia -> Ruditapes_philippinarum -> .. -> Spio_sp の順に、
Ako-Nisio間の非類似度への寄与が大きいことがわかります。
#32-33 simper() の計算結果を確認します。
コード#31 では、累積寄与度の結果のみが出力されますが、実際にはたくさんの計算結果(詳細は省略)が保存されています。ひとまず simper() の計算結果を sim という箱に格納することで、あとから全ての計算結果を確認することができます。
※注意1
simper()は、グループデータがカテゴリカルでないと(=数値データが入力されると)実行されません。
※注意2
出力される寄与度は「累積寄与度」です。2番目以降に出現するカテゴリーの寄与度を知るためには、以前の寄与度を引く必要があります。
たとえば…
Ruditapes_philippinarum の寄与度は 0.4156330 - 0.3128506 = 0.1027824となり、
Hima_festiva の寄与度は 0.4920827 - (0.3128506 + 0.4156330) = … となります。
ほかにも色々な統計解析方法があるのですが、とりあえず今回はここまでとさせていただきます。
(リクエストがあればコメントください!)
プロットをカスタマイズする
ここからは、nMDS で得られた2次元プロットをカスタマイズするためのコードを紹介します。
色と形を変更する
fauna データを水域 [Ako, Nisio, Nakatu, Banzu, Uto] ごとに異なる色と形を与えてプロットしたいとします。
cols <- c("#ce6066","#987f47","#6b8c41","#5f80cd","#996dd3")
shape <- c(1,15,16,17,18)
site_n <- as.numeric(as.factor(site))
ordiplot(bcmds,type="n")
points(bcmds, col=cols[site_n], pch=shape[site_n], display="sites")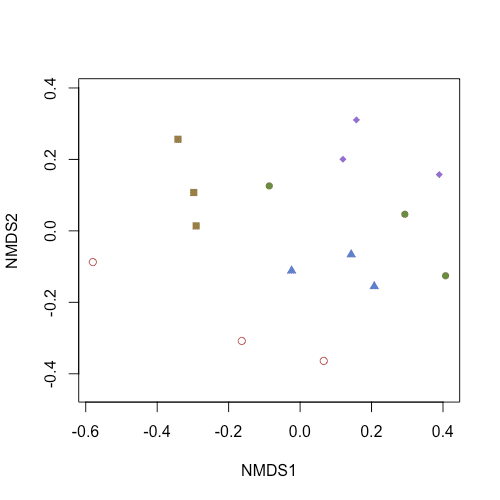
解説
#34 色の情報が格納されたデータリストを作成します。
#35 形の情報が格納されたデータリストを作成します。
#36 後のプロット作成の都合で、#24-28で作成した水域IDが格納されたデータリスト "site" に少し修正を加えます。
siteに格納されているcharacter型のデータをnumeric型に変更します。as.numeric()関数でデータ型を numeric型 に変更できるのですが、Rでは character型 -> numeric型 へ直接変更することができません。このため、中間に factor型 をかましています。
(書いている途中で型がゲシュタルト崩壊しました)
#37 ordiplot() で "キャンバス"を用意します。 (#7,10,15と同じ)
#38 points() でデータをプロットします。 (#8,11,16と同じ)
Rのplot()関数と同様に、col= と pch= でプロットの色と形を指定できます。
「fauna データを水域 [Ako, Nisio, Nakatu, Banzu, Uto] ごとに異なる色と形を与えてプロット」するために、site (site_n) に格納されているIDと対応させて色や形の情報を与える必要があります。
col=cols[site_n], pch=shape[site_n] とすることで、site "Ako" に 色 "#ce6066"と形 "1"を、 site "Nisio" に色 "#987f47"と形 "15" … を与えられます。
なお、cols[], shape[] の []の中身が numeric型でないと、このコードは機能しません。そういうわけで、character型のsiteデータを numeric型に変換しました (#36のコード)。
凡例をつける
どの水域にどの色と形のプロットが対応しているか、一目でわかるように凡例を載せます。
ordiplot(bcmds,type="n")
points(bcmds, col=cols[site_n], pch=shape[site_n], display="sites")
legend("bottomleft",
legend=c(unique(site)),
col=cols[(unique(site_n))],
pch=shape[unique(site_n)])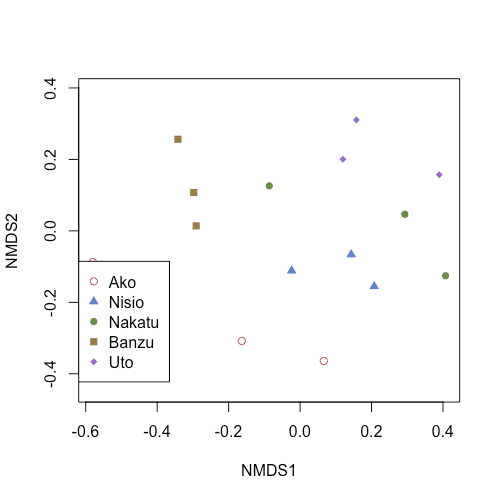
解説
#39 #37と同じ
#40 #38と同じ
#41-44 legend() で凡例を出力します。
"'bottomleft"で、凡例の位置を指定します。topright, topleft, bottomright などがあります。
legend= で水域のID、col=で各IDの色、pch= で各IDの形を指定します。
legend=ではcharacter型の入力が許されるためsiteを指定しました。一方、col=やpch=ではnumeric型データであるsite_nを指定します。理由は先に述べた通りです。
なお、unique() は、指定のデータリストから重複しないように要素を抽出するための関数です。veganに限らず、Rを使ったデータ処理ではよく登場します。
unique() を指定しないとsite に格納されている全データが引っ張りだされるので、Ako, Ako, Ako, Nisio, Nisio, Nisio, Nakatu … (ここで途切れる) みたいな凡例ができあがります。
高解像度で図を保存する
最後に、出力された図を高解像度で保存するコードを紹介します。
png("nMDS.png", width = 1200, height = 1200, res=200)
ordiplot(bcmds,type="n")
points(bcmds, pch=shape[site_n], display="sites", col=cols[site_n])
dev.off()プロットの前後でpng()とdev.off()を実行すれば、現在のディレクトリに図が保存されます。
res=の数値を大きくするほど解像度があがります。論文の図を作成するときには、この方法での保存がおすすめです。
長くなりましたが、今回はここまでです。
この記事ではnMDSを紹介しましたが、そのうち他の次元圧縮方法を紹介するかもしれません。脂肪酸組成データの解析方法も紹介したいですね・・・。
追記
nMDSのプロットに楕円を表示する (20240123)
コード#34-38で得られた図に、95%信頼楕円を重ね書きするコードを紹介します。
cols <- c("#ce6066","#987f47","#6b8c41","#5f80cd","#996dd3")
shape <- c(1,15,16,17,18)
site_n <- as.numeric(as.factor(site))
ordiplot(bcmds,type="n")
points(bcmds, col=cols[site_n], pch=shape[site_n], display="sites")
### ここから追加 ###
ordiellipse(bcmds, groups = site_n,
display="sites",
kind="sd",
conf=0.95,
label=FALSE,
col=cols,
draw="polygon",
alpha=100,
border=FALSE)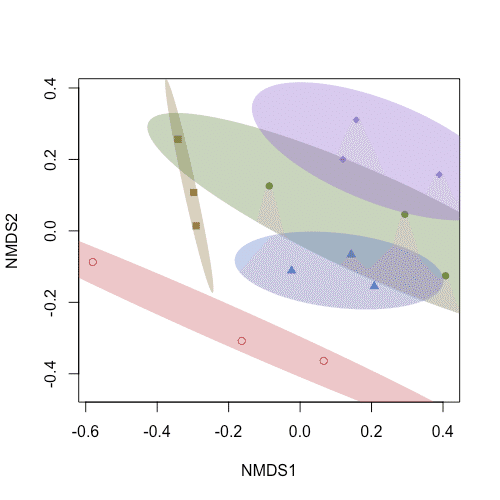
解説
#45 ordiellipse() で、楕円を表示します。
以下、ordiellipse() の引数について、重要な点のみを解説します。
復習ですが、bcmdsは nMDSの計算結果でした(コード#5参照)。
group = で、グループ分けの基準とするデータ列を指定します。
今回は、水域 [Ako, Nisio, Nakatu, Banzu, Uto] ごとに異なる色でプロットしたので、楕円もこの基準にそって色分けしましょう。
よって、group = site_n としました。
kind = で楕円面積の計算方法を選択します。選択肢は、"se", "sd", "ehull" があります。
conf = で楕円の信頼区間を設定します。よく使われるのは95%信頼区間だと思うので、conf = 0.95としました。
col = でカラーパレットを指定します。今回は、コード#34で作成したカラーパレットを活用します。
draw = で楕円の描画方法を設定します。楕円を塗りつぶしたい場合は"polygon"、楕円の線だけ表示したい場合は"lines"としてください。
alpha = で楕円の透明度を設定します。値が小さいほど、色が薄くなります。
※今回のサンプルデータセットは、サンプルサイズが小さいため、楕円の面積がかなり大きくなります。
楕円が切れて気になる場合は、ordiplot() 内で、xlim と ylim を設定すると良いですよ。
例)
ordiplot(bcmds,type="n", xlim = c(-1,1), ylim = c(-1,1))
points(bcmds, col=cols[site_n], pch=shape[site_n], display="sites")
ordiellipse(bcmds, groups = site_n,
display="sites",
kind="sd",
conf=0.95,
label=FALSE,
col=cols,
draw="polygon",
alpha=100,
border=FALSE)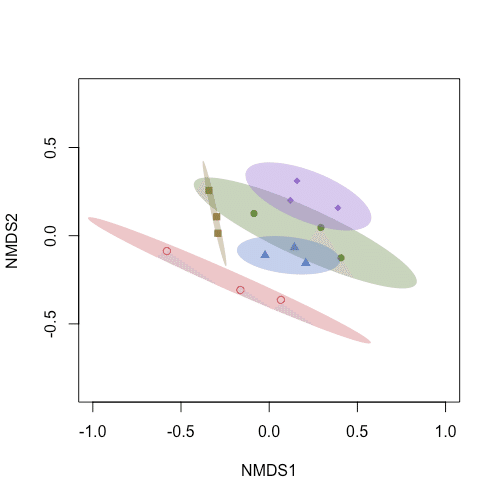
また、ベクターを同時に表示したい場合は、最後に該当コードを実行すればOKです。
ordiplot(bcmds,type="n", xlim = c(-1,1), ylim = c(-1,1))
points(bcmds, col=cols[site_n], pch=shape[site_n], display="sites")
ordiellipse(bcmds, groups = site_n,
display="sites",
kind="sd",
conf=0.95,
label=FALSE,
col=cols,
draw="polygon",
alpha=100,
border=FALSE)
plot(envNMDS, col="black")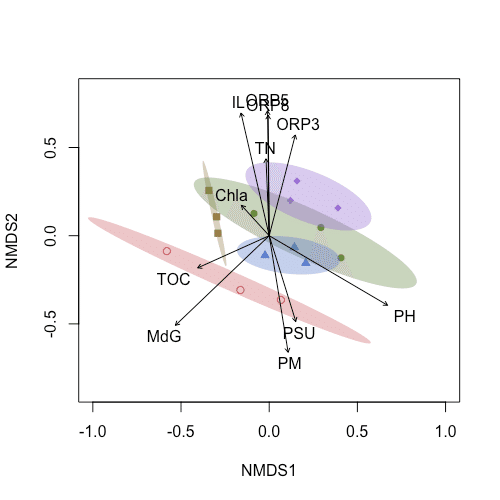
※ plot(envNMDS, col="black") については、コード#13-17参照
おわり