
【W4】リガンドベーススクリーニング_09_余談

【類縁体選抜の課題】
前回でTeachOpenCADDのW4に関しては体験を完了しています。今回は徒然なるままに類縁体選抜について心にうつりゆくよしなしごとを、そこはかとなく書きつけてみます。よろしければおつきあいください。
W4では化合物をエンコード(記述子、フィンガープリント)し、比較(類似性評価)する様々なアプローチを取り扱いました。さらに、バーチャルスクリーニングを実施しました。
フィンガープリント2種、類似度指標2種の各組み合わせを指標に化合物の類似度が高い順に化合物を選抜するバーチャルスクリーニングを体験しました。
確かに性能差はありました。一方で、それほど劇的な性能向上もなかったと思いませんか。
他にも下記のような課題もあると思います。
化合物フィンガープリントを使用した類似度検索の欠点は、化合物類似度に基づくものなので新規な構造を生み出さないことです。化合物類似度を扱う上でのその他の課題としては、いわゆるアクティビティクリフ(activity clliff)があります。分子の官能基におけるちょっとした変化が生理活性の大きな変化を起こすことがあります。
(引用元)
【似ていても 意外と小さい タニモト係数】
実のところ、類似度をしきい値にして化合物の絞込みって創薬化学者が「似ている」と感じる感覚とずれていることがあります。
上記記事がとても具体的な例を挙げて類似度について考察して下さっています。一部引用させていただきます。
用いるフィンガープリントはmorganのradius=2, bitInfo=2048という計算条件です。タニモト係数で評価しています。
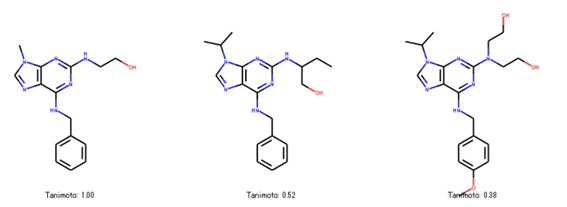
タニモト係数は順に1.00, 0.52, 0.38という結果でした。構造を見た感じではmol2(真ん中)、mol3(右)ともにmol1(左)とかなり類似していますが、タニモト係数は結構小さいなという印象を受けました。
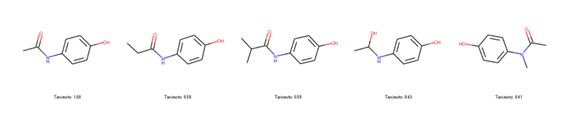
タニモト係数は順に1.00, 0.59, 0.59, 0.43, 0.41という結果でした。メチル基を側鎖に1つ追加することで、タニモト係数が1→0.59に変化することが分かりました。(感覚的には0.59よりもっと大きな値になるイメージでした)
【創薬化学者の類縁体選抜手法の例】
創薬化学者は化合物を比較する際、「母核」とか「共通骨格」と呼ばれる部分構造の類似性を重視する傾向があり、「側鎖」とか「置換基」の変化があっても「母核」が一致すれば類縁と見なすことは多いです。
ただしこの「母核」の定義は人によっても目的によってもまちまちです。
いかにプログラム上で「共通骨格」を定義し、化合物群の解析に利用するかはTeachOpenCADDだとW6で扱っています。MCS (Maximum common substructures)に注目した解析方法を体験する予定です。
さて、コンピューターではなくヒトすなわち今回だと創薬化学者は部分構造に注目した類縁体選抜をどのように行うでしょうか?
デモデータを使ってKNIMEも利用しながら体験してみます。
既知のEGFR阻害剤ゲフィチニブ(Gefitinib)をクエリとして使用し、EGFRに対して試験済みの化合物データセットの中から類似した化合物を検索します。
(引用元) https://magattaca.hatenablog.com/entry/2020/04/19/223829
【KNIMEでの部分構造検索】
データセットは4510化合物として、ゲフィチニブに類似した化合物を例えば100個選抜してと要求されたとします。実際には「最小活性発現構造」とか「独自性のある新規スキャフォールド」とか「sp3性向上」とか特殊な目的も加わるので単純ではないのですが、創薬化学者はおそらく最初は4510化合物に対して何種かのQuery(検索式)を立てて部分構造検索して化合物リストを数百まで絞ってから目視で選抜するのではないでしょうか?違っていたらすみません。しかし上記の仮定で進めます。
さて、部分構造検索ですが二通り紹介済みです
私はSMARTSをスマートに使いこなせない上にJChem推しなので再びMarvin Sketchを使います。
用いるデータはW4のメタノードと同様にW2の出力を利用します。
Marvin SketchとRDKit Molecule Substructure Filterの2つを繋ぎます。
【Marvin Sketch】
前の記事と設定は変わりません。
構造Queryの入力については少しだけ新しい話をします。
Name To Structure機能を利用します。
こちらの記事はプロが書いているので内容が濃いです。一読をお勧めします。Document to Structureはあいにくトライアルライセンスをもらわなかったのでそのまま真似は出来ないのですが、幸いにして化合物名から化学構造を認識するName to Structureの英語版に限ってはMarvinのトライアルライセンスでも利用できます。早速使って見ましょう。
A)文字列” Gefitinib”をコピー(例:Gefitinibを選択して「Ctrl+C」)。
B)MarvinSketchノードを開く
C)描画ウィンドウ内で「Crtl+V」などしてペースト
D)描画された構造を確認、必要に応じて編集
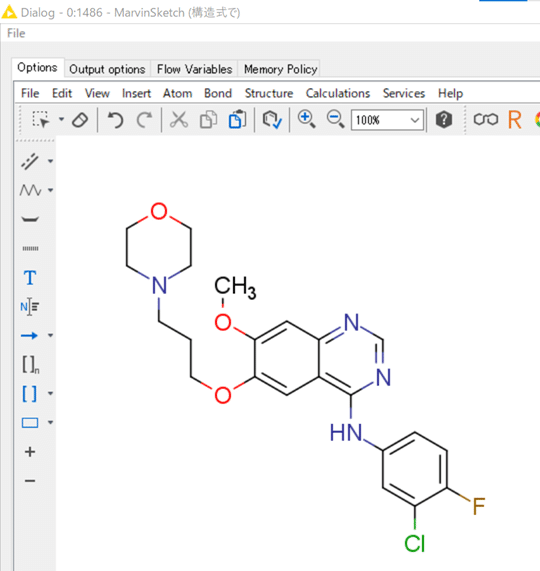
便利ですよね!
ここまではいいんですが、実は今回も正しくRDKit Molecule Substructure Filterで検索できるように、芳香環上のボンドは全てAromaticに変える(すなわちアロマタイズする)必要があります。これは手間が増えてしまうので残念なところ。
JChem ExtensionsのMol Searchノードに繋ぐならこの操作はいらないと思うのですが、これまたトライアルライセンスがなくて使えないので断念しました。検索リストで分子をそろえて表示してくれたりと便利なんですよ!
今回用いる検索式は下図の通りアロマタイズしておきました。
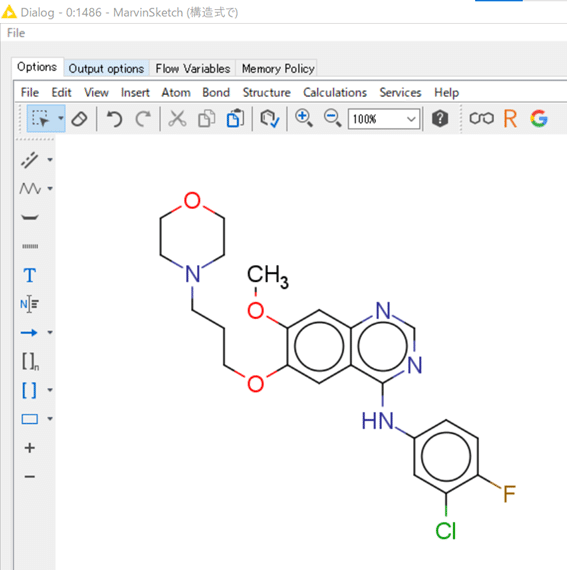
アロマタイズってアロマタイムと一字しか違わないんですね。ほっこりします。
【RDKit Molecule Substructure Filter】
こちらも先日の記事の通りの設定です。
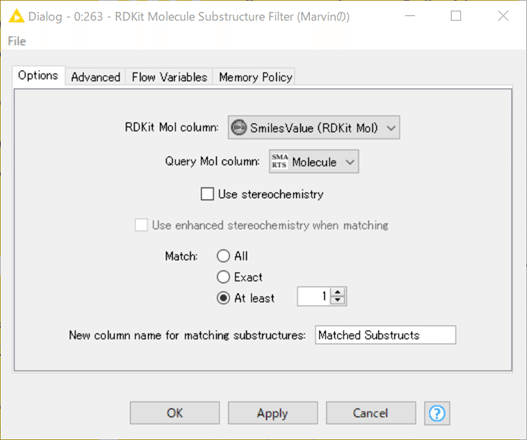
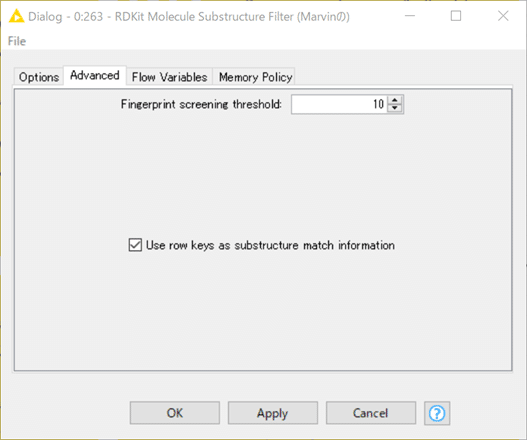
結果:
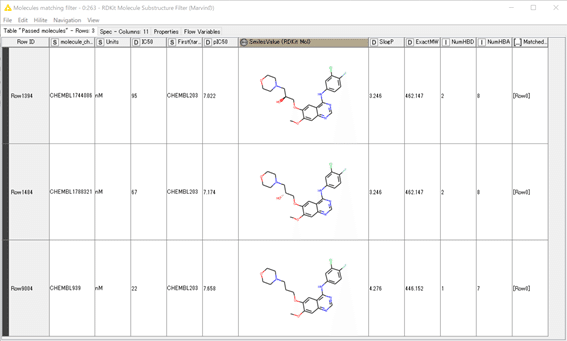
側鎖に水酸基が生えた化合物もあるのですね。
【Queryを工夫して追加で検索】
今回は、Gefitinibの側鎖を削っても活性が残るかを調べる化合物セットでも探そうかと思いますので、下記のQueryで再検索してみます。
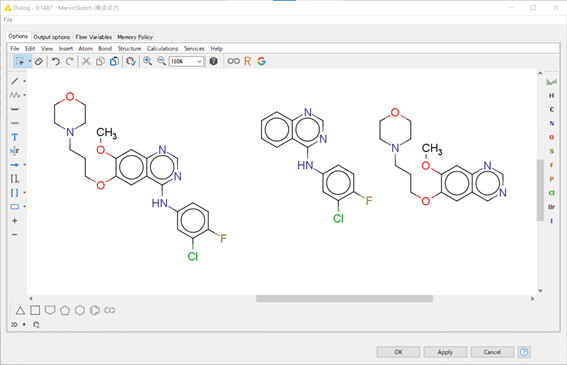
結果:
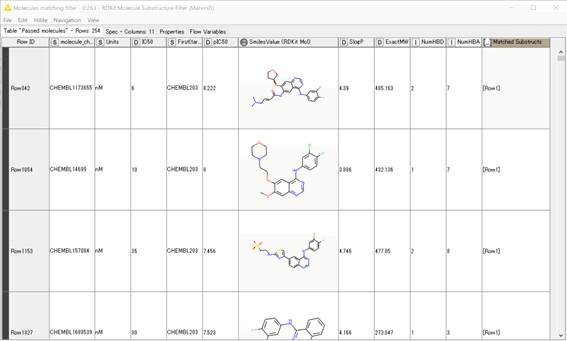
254化合物がヒットしてきました。
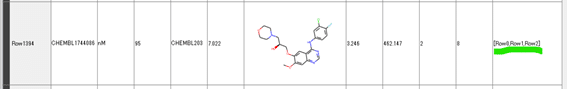
複数のQueryでヒットした場合は、QueryのIDリストで出力してくれる親切設計です。
創薬化学者の方が手作業で調べる場合は3つそれぞれのQueryで調べて、それぞれSDFとかExcelに出力して、重複チェックは紙に印刷してペンで書きこみながらやったりしてませんか?さすがに2020年代だと印刷まではしないか??
KNIMEだとデータの集計や加工はお手のものなので一度お試しいただけたらなと思います。
ちなみに上記の254化合物のリストの場合活性値もついているので、もし仮にこれらの化合物をスクリーニングにかけたらどれぐらいのヒット率かなどのシミュレーションもできます。自分なりにいろいろ絞り込みをしてみて、答え合わせをして楽しめます。奇特すぎるか。
Structure To NameでGefitinib のIUPAC名を見ると
N‐(3‐chloro‐4‐fluorophenyl)‐7‐methoxy‐6‐[3‐(morpholin‐4‐yl)propoxy] quinazolin‐4‐amine
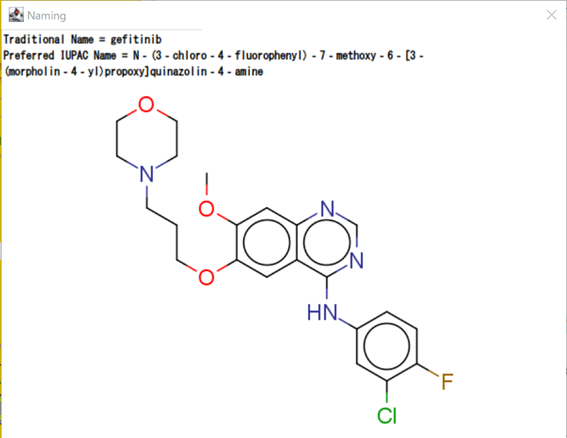
なんですが、キナゾリン環の置換基として4位のアリールアミノ基だけあれば活性はある程度(IC50=30nM)ありますね。
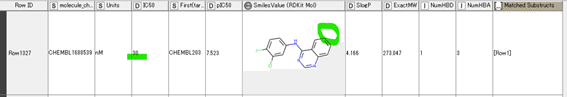
あるいはこんな化合物もヒット(IC50=20.7nM)するんですね。
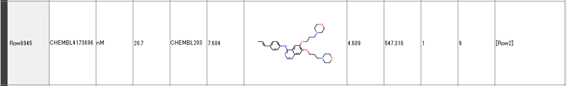
とつい楽しんでしまいました。あやしうこそものぐるおしけれ。
余談もそこそこで切り上げることにします。次回からはW5へ進めます。
いいなと思ったら応援しよう!
