高分子物性計算の自動化を支援するPythonライブラリ RadonPyのセットアップと動作テスト

概要
RadonPyは、高分子物性計算の自動化を支援するPythonライブラリです。力場計算などを自動で行ってくれるので、シミュレーションの専門家でなくても動かせるようです。
最近は富岳を使った大規模計算のプロジェクトが走っています。
自分でも、触ろう触ろう思っていたのですが、本日、思い立って動かしてみることにしました。
ポリスチレンの物性を計算するサンプルコードを回します。
立ち上げ
使用環境は以下の通り。
Ubuntu 22.04.3 LTS
AMD Ryzen Threadripper 3990X 64-Core Processor
公式マニュアルに従ってコマンドを打っていきます。
conda環境の作成
conda create -n radonpy python=3.11
conda activate radonpy関連ライブラリのインストール
condaのconflictが発生したので、pipと組み合わせたりしてinstallしました。
#lammps
conda install-c conda-forge lammps
#psi4
conda install -c conda-forge/label/libint_dev -c conda-forge -c psi4 psi4 resp
conda install -c conda-forge/label/libint_dev -c conda-forge -c psi4 resp
#rdkitなど
pip install rdkit-pypi
pip install mdtraj
pip install matplotlib
pip install radonpy-pypi
#DFTのd3モジュールも必要でした
conda install -c psi4 dftd3参考: 自分の環境では上手くいかなかった例
全てを一気にcondaで入れる: moduleのconflictが発生
conda install -c conda-forge/label/libint_dev -c conda-forge -c psi4 rdkit psi4 resp mdtraj matplotlib2. pipでlammpsを入れる: lammpsの並列計算(lmp_mpi)機能が入っていない
pip install radonpy-pypi[lammps]ポリスチレンの計算
チュートリアルのコードを、VS codeで動かしてみます。
諸々の初期化と初期設定
from radonpy.core import utils, poly
from radonpy.ff.gaff2_mod import GAFF2_mod
from radonpy.sim import qm
from radonpy.sim.preset import eq, tc
smiles = '*C(C*)c1ccccc1'
ter_smiles = '*C'
temp = 300
press = 1.0
omp_psi4 = 10
mpi = 10
omp = 1
gpu = 0
mem = 10000
work_dir = './work_dir'
ff = GAFF2_mod()molオブジェクトの生成
mol = utils.mol_from_smiles(smiles)notebook上でmolと実行すると、分子構造が表示されます。
mol
変数のタイプを確認すると、RDKitのmolオブジェクトのようです。
結合部分は3H(トリチウム)などでラベリングしている印象を受けます。
計算にはwork_dirを作っていく必要があるようなので、作ります。
import os
os.mkdir("work_dir")安定構造の計算
mol, energy = qm.conformation_search(mol,ff=ff,work_dir=work_dir,
psi4_omp=omp_psi4,mpi=mpi,omp=omp,memory=mem,log_name='monomer1')1 min程度で計算が終わりました。
電子物性の計算
# Electronic propety calculation
qm.assign_charges(mol,charge='RESP',opt=False,work_dir=work_dir,
omp=omp_psi4,
memory=mem,log_name='monomer1')
qm_data = qm.sp_prop(mol,
opt=False,work_dir=work_dir,
omp=omp_psi4,memory=mem,log_name='monomer1')
polar_data = qm.polarizability(mol,opt=False,work_dir=work_dir,
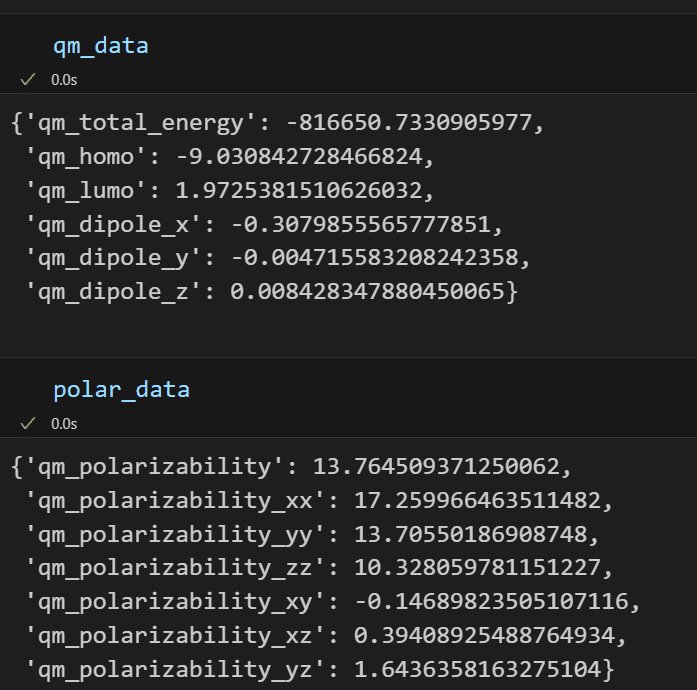
omp=omp_psi4,memory=mem,log_name='monomer1')計算結果は以下の通り。物性を計算できてますね。
末端に関するRESP chargeの計算
# RESP charge calculation of a termination unit
ter = utils.mol_from_smiles(ter_smiles)
qm.assign_charges(ter,charge='RESP',opt=True,work_dir=work_dir,omp=omp_psi4,memory=mem,log_name='ter1')高分子構造の生成
# Generate polymer chain
dp = poly.calc_n_from_num_atoms(mol, 1000,terminal1=ter)
homopoly = poly.polymerize_rw(mol, dp,tacticity='atactic')
homopoly = poly.terminate_rw(homopoly, ter)2minほどかかりました。分子量が1000になるように、重合度(dp)を決めるようです。

以下、本格的なMD計算となるようです。
待ち時間が長くなります。
MD計算(平衡化処理)
# Force field assignment
result = ff.ff_assign(homopoly)
if not result:
print('[ERROR: Can not assign force field parameters.]')
# Generate simulation cell
ac = poly.amorphous_cell(homopoly, 10,density=0.05)
# Equilibration MD
eqmd = eq.EQ21step(ac,work_dir=work_dir)
ac = eqmd.exec(temp=temp,press=1.0,mpi=mpi,omp=omp,gpu=gpu)
analy = eqmd.analyze()
prop_data = analy.get_all_prop(temp=temp,press=1.0,save=True)
result = analy.check_eq()追加の平衡化計算と熱伝導度の計算
# Additional equilibration MD
for i in range(4):
if result: break
eqmd = eq.Additional(ac,work_dir=work_dir)
ac = eqmd.exec(temp=temp,press=press,mpi=mpi,omp=omp,gpu=gpu)
analy = eqmd.analyze()
prop_data = analy.get_all_prop(temp=temp,press=press,save=True)
result = analy.check_eq()
if not result:
print('[ERROR: Did not reach an equilibrium state.]')
# Non-equilibrium MD for thermal condultivity
else:
nemd = tc.NEMD_MP(ac,work_dir=work_dir)
ac = nemd.exec(decomp=True,temp=temp,mpi=mpi,omp=omp,gpu=gpu)
nemd_analy = nemd.analyze()
TC = nemd_analy.calc_tc(decomp=True,save=True)
if not nemd_analy.Tgrad_data['Tgrad_check']:
print('[ERROR: Low linearity of temperature gradient.]')
print('Thermal conductivity: %f' % TC)一晩くらいは計算に時間がかかりそうです。
計算が終わったら、結果を追記するかもしれません。
結果(追記)
2日ほど放置すると、計算が終わっていました(正確にはerrorで終了)
一部の物性をうまく計算できなかった印象です。

ただ、work_dir/analyze内に、一部の計算結果は保存されていました。
例えば密度の計算値は0.99 g/cm3で、実測(1.05程度)と概ね一致しました。
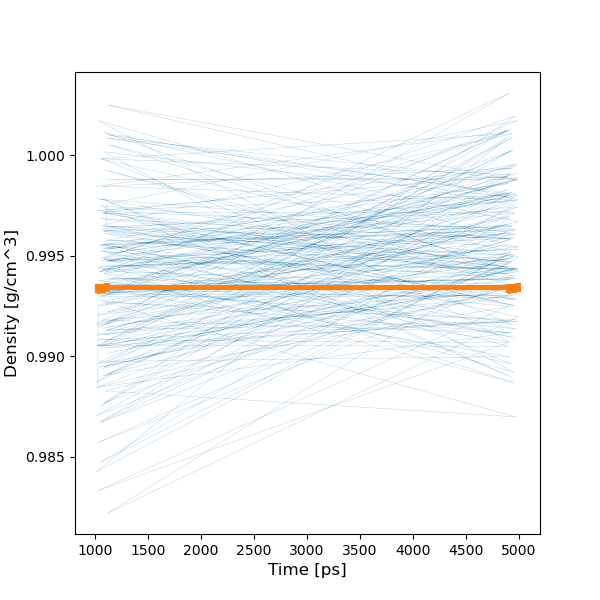
今後はエラー内容の解析・対策や、計算途中からの復帰法などを、理解する必要がありそうです。
まとめと感想
MD計算システム、RadonPyを立ち上げてみました。
MD計算の鬼門といえる、力場計算を全自動でできるのは、素晴らしいです
精度や信頼性については(特にMDの専門家からすると)、議論がありそうですが、機械学習モデルの入力変数としての活用など、色々と用途が広がりそうです