
先天性QT延長症候群・メモ(2021年版)

江花有亮
東京医科歯科大学病院遺伝子診療科
2021年5月6日初稿
0.はじめに
先天性QT延長症候群は心臓に構造的な異常を認められないにもかかわらず突然死のリスクが高い遺伝性不整脈の一つです。QTが延長する原因は様々ですが、心筋細胞の脱分極や再分極の調整に関与するイオンチャネルやそのチャネルの機能を支えるタンパク質をコードしているいくつかの遺伝子の変異に起因しております。QT延長症候群の患者の症状は無症状から突然死まで幅広く見られます。一般的に、小学校あるいは中学校健診の心電図で指摘を受けることが多いですが、まれに突然死が初発の症状であることもあるますため、生活管理や正確な診断が非常に重要です。
本書では2018年に改訂された日本循環器学会「遺伝性不整脈の診療に関するガイドライン」や2013年に米国・欧州・アジア不整脈学会(HRS/EHRA/APHRS)から出版されたHRS/EHRA/APHRS Expert Consensus Statement on the Diagnosis and Management of Patients with Inherited Primary Arrhythmia Syndromeを解説し、またこれらガイドライン出版後に示されたエビデンスをもとにまとめております。
本書が想定している読者は先天性QT延長症候群に関わる人たちです。
1.QTとは何か
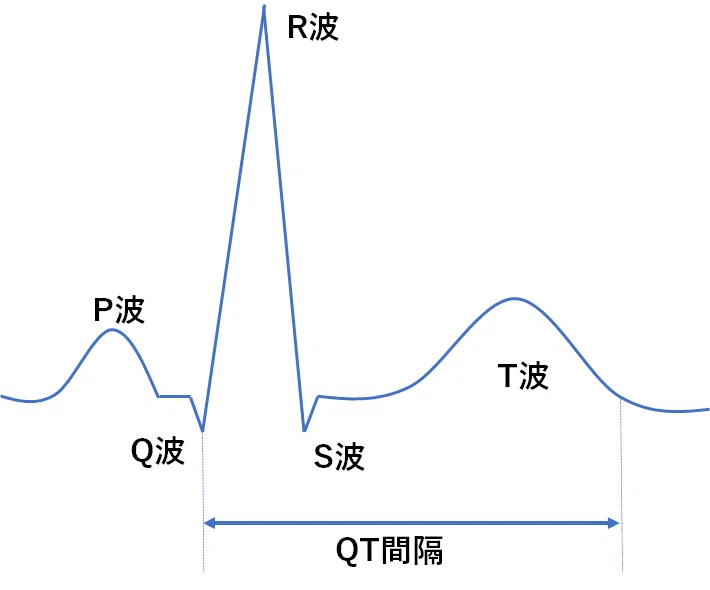
QTとは心電図波形上の特徴的な所見から「P波」(PulseのP)「Q波」「R波」「S波」「T波」「U波」と命名されており、QTとは「Q波」から「T波」までの長さを表す言葉である。心臓の構造は4つの腔から成っており、それぞれ右房、左房、右室、左室である。心臓刺激伝達系については動画参照。右房上部の存在するペースメーカー細胞から生成された電気的興奮が右房から左房へ伝達する過程でみられるのが「P波」である。そして電気的興奮が房室結節を経て右室・左室の伝導路と通じて電気的興奮が伝わることで右室・左室が収縮する。左室の収縮によって心臓のポンプとしての機能を果たして全身へ血液が送られる。左室の収縮によって全身から戻ってきた血液を酸素化するために肺へ送られる。右室と左室、両心室へ送られる過程は心電図上Q波からR波を経てS波へ至る「QRS」によって表される。そして最後に収縮した両心室が弛緩する過程でみられるのが「T波」である。「QT間隔」とはQ波からT波の終わりまでの間隔を指し、それは両心室の収縮から弛緩までの過程全体を表している。
一般的には心拍数によって補正QT(QTc; corrected QT)が用いられる。Bazettの式、QTc=QT×√RRで示される。RRととなりある心拍のR波からR波の間を指し、心拍数が60拍/分であればRR=1秒となる。

2.QT延長の意味とは
QT延長とは両心室の収縮の過程と弛緩の過程を表すのであるが、先天性QT延長症候群と診断された症例の半数以上では弛緩の過程が延長している。心室の弛緩の過程は心室性不整脈の影響を受けやすい不安定な部分である。QTが延長するということはその不安定な状態が長く続くことで、当たり所が悪く不整脈が出現したときに、致死性の不整脈が起こってしまう可能性が高いという事である。QT延長は様々な状況で起こる。
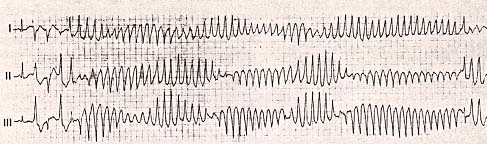
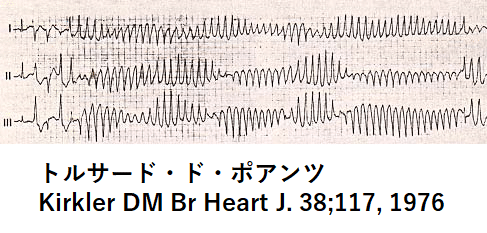
3.心イベント、および致死性不整脈とは
先天性QT延長症候群では心イベントがどれくらい起こりやすいのかが重要である。心イベントとは失神と致死性不整脈、突然死が挙げられる。起こりやすい致死性不整脈として、心室細動とトルサード・ド・ポアンツという不整脈が挙げられる。心電図上の特徴としてギザギザの鋸の歯のような形状の波形がみられる。ここまでランダムで、非常に速い心拍では全身に血流を送るポンプの機能を果たすことができない。脳への血流の減るか途絶えてしまうので失神という症状で現れ、その不整脈が長く続けば脳のみならず全身への血流が保つことができなくなるため死に至る。
先天性QT延長症候群に限らず、遺伝性不整脈は長期にわたり無症状のこともあるが、心室頻拍(VT)、心室細動(VF)などの致死性不整脈を発症するとまず動悸、めまい、失神などの症状が出現した後、その状態が長く続くと心停止・心臓突然死の原因となりうる。
人口動態統計での不整脈死の割合と心臓突然死の頻度
心臓突然死は心臓に起因する予期せぬ内因死である。2019年(令和元年)の人口動態統計によると、心疾患による死亡数は約20.7万人で、全死因の約15.0%である。そのうち不整脈及び伝導障害による死は約3万人で2.3%を占めていた。また乳児死亡数892人のうち乳幼児突然死症候群は4.8%であった。
突然死の年間発生率は久山町研究では40歳以上の住民10万人あたり89人、京都市民の調査では10万人当たり102人、新潟県の15歳以上の調査では10万人当たり145人であった。日本人においては10万人当たり100人程度と推測され、年間13万人が突然死していると考えられる。
久山町研究で、1962~2009年に剖検を施行した1,934人のうち発症から24時間以内の突然死は204人、そのうち心臓突然死は100人であった。そのうち23%は原因不明の心疾患であり、このような原因不明の心疾患の一部は遺伝性不整脈であった可能性がある。通常の病理解剖のうち、器質的心疾患を認めなかった突然死症例に対し、遺伝子検査を実施したところ、17.6~26.1%に遺伝性不整脈の遺伝子変異が認められた。
日本の状況をまとめると、わが国では年間約13万人の突然死の発生があり、そのうち約7万人が心臓突然死であり、遺伝性不整脈はそのうちの約10%、7,000人程度と考えられる。
一般的な診断の流れ
失神や心停止などの重篤な症状を認めた場合、病歴や家族歴、検診所見などとともに、一般的に心臓超音波検査、心臓MRI、冠動脈造影検査などの画像検査が行われる。同時に12誘導心電図、24時間ホルター心電図検査などを合わせて行う。あるいは二次性不整脈の原因となるような、薬物、電解質異常、代謝性疾患、外傷などを調べる。これらの検査で心臓その他に明らかな器質的異常(=構造的な異常)を認めない場合、遺伝性不整脈が疑われる。
遺伝性不整脈が疑われる場合、入院あるいは外来においてカテコラミンやNaチャネル遮断薬などの負荷試験や心臓電気生理検査によるリスク判定を目的として適応が検討される。
4.先天性QT延長症候群の疫学
先天性QT延長症候群の有病率はこれまで2,000人に1人の割合で認められ、やや女性に多い 。初回心臓イベント(失神、心室細動、心停止、突然死)の平均発症年齢は男性で8歳、女性で14歳である。初発症状として致死性イベントは5%未満に認められる。
QT延長そのものは病的な遺伝子変異を伴う先天性QT延長症候群でなくても、上記のような薬剤性や心疾患の合併等でも起こる。QT延長症候群の診断基準に照らして3.5点以上で診断確実となるが、近年では遺伝子診断の結果を踏まえて診断することが多くなっている。
有病率
イタリアで行われた前向き研究で、15~25日齢の44,596人の白人乳児のECGを調査し、繰り返しQTc > 450msを認めた症例について遺伝子解析スクリーニングをした。①470msを超える乳児31人、②461~470msの乳児28人、③451~460msの乳児184人認めた。①について31人中28人遺伝子解析をして12人に、②について28人中14人検査をして4人が変異陽性であった。結果として2,000人に1人の割合と考えられている。
わが国では乳児1か月検診に合わせた心電図所見に基づいた疫学調査を実施しており、QTc>]470msを継続的に示す児は4,285人中4人(0.09%、約1,100人に1人の割合)であった。1994年から実施されている心電図健診から、遺伝的検査を含まないリスクスコアによる診断基準(Schwartzの旧基準)では中学1年で有病率が高く、1,200人に1人であった。
年齢
先天性QT延長症候群の発症年齢、すなわち初回心イベント時の年齢は乳幼児から成人期まで広く分布する。学童期から思春期の発症が多い。平均発症年齢は男性8歳、女性14歳である。初回心イベント全体の90%は40歳までに見られる。
男性発端者で、15歳以降に初回心イベントを認める症例は8%にとどまるが、女性発端者では40%である。
小児先天性QT延長症候群患者3,015人のレジストリーによれば、1~12歳の観察期間に心停止又は突然死を来す率は男児で5%、女児で1%であり、男児の方が高い。13~20歳では性差なし、18~40歳では女性が男性の3倍の割合である。
性差
先天性QT延長症候群はやや女性に多い傾向がある。先天性QT延長症候群の原因となる親の変異アレル(ヒトであれば2本ある遺伝子座のうちの片方)は男児よりも女児に遺伝されやすく、特に母親から女児へ遺伝する率が高いという報告がある。この点が、先天性QT延長症候群が女性に多く見られることの一因と推定されている。
家族歴
ほとんどは常染色体優性(顕性)遺伝形式である。Jervell and Lange-Nielsen症候群などの稀なケースでのみ常染色体劣性(潜性)遺伝形式となる。遺伝子変異が認められた先天性QT延長症候群患者の85%は前世代から遺伝したものであったが、15%が新規発生(de novo)の変異であった。
遺伝子変異は確率的には50%で次世代へ遺伝するが、先天性QT延長症候群患者1,534人の検討では次世代へ870人(57%)が同一遺伝子変異キャリアであり、664人(43%)が非キャリアであったという報告がある。また
5.QT延長の原因となる状況
先天性QT延長症候群のほかに薬剤や徐脈などが原因で二次的にQT延長が起こり、致死性不整脈が発生することがある。一番多いのはある種の薬剤を服用しているときであり(BOX1)、抗不整脈薬による致死性不整脈発現の頻度は2.0~8.8%と考えられている。その他、向精神薬、抗生物質、抗真菌薬、抗アレルギー薬、消化器疾患薬でもQT延長を来す。抗不整脈薬以外の薬剤によるQT延長の頻度は10,000~100,000人に1人と考えられている。
心不全や心筋症、冠動脈疾患、高血圧、左室肥大などの基礎心疾患の合併によりQT延長が助長される。また洞不全症候群や房室ブロックなどによって徐脈となると著名なQT延長を来し致死性不整脈が発生する例がある。
その他、高齢、女性、脱水、電解質異常(特に低カリウム血症、低マグネシウム血症)、更に糖尿病やアルコール中毒、神経性食思不振症、下垂体・副腎不全、甲状腺機能低下症、低体温、低血糖などの代謝障害によるQT延長がある。クモ膜下出血、頭蓋内出欠の急性期にもQT延長を認める。
薬剤
二次性QT延長症候群のうち最も頻度が高いものが薬剤によるものである。その影響は古くから指摘されており、循環器で使用する薬剤以外にも多くQT延長作用を持つ薬剤が存在する。薬剤開発の際には必ずQT延長作用についてチェックされる。QT延長をきたしうる薬剤についてはCredibleMedsというウェブサイトが便利である。日々、最新の情報がアップデートされている。抗アレルギー薬や向精神薬などQT延長作用の強い薬剤が多かったが、現在ではQT延長作用の少ない薬剤が開発され、いくつかは発売中止となっている。

薬物相互作用
抗不整脈薬は肝臓においてチトクロムP450という酵素によって代謝を受け分解される。主なものはCYP2D6とCYP3A4という代謝酵素である。
中にはこれらCYP2D6やCYP3A4の働きを阻害する(邪魔をする)薬剤があり、これら薬剤を併用すると抗不整脈薬が分解されず長く体内に残存し、血中濃度が上昇してしまう。パロキセチン(パキシル®)やシメチジン(タガメット®)、チオリダジン(メレリル®2005年販売中止)はCYP2D6の酵素阻害を起こす。一方、エリスロマイシン、クラリスロマイシン、フルコナゾール、ジルチアゼムはCYP3A4の酵素阻害を起こす。
(1) 抗不整脈薬
不整脈の薬は薬効によって大きくIからIVに分類されている。I群はナトリウムチャネル遮断薬であり、その一つであるキニジンによるQT延長とトルサード・ド・ポアンツは「キニジン失神」とも言われている。I群の中でもIa群やIc群は心房細動などの心房性不整脈に使用されるケースが多い。心房細動は日常診療において多く見られる不整脈であるが、QT延長を伴う場合にはカテーテル・アブレーションや別の薬剤への変更などを考慮する。
III群も心房細動などの心房性不整脈でも用いられるが、致死性の高い心室性不整脈でも用いられるケースがあるので慎重に決定する。
これら抗不整脈薬によるトルサード・ド・ポアンツTdPの発生率は有意に女性が多い。これら患者の64-75%は女性である。
(2) 抗精神病薬、抗うつ薬
向精神薬
QT延長の多い薬剤である。エスシタロプラム(レクサプロ®)、クロミプラミン(アナフラニール®)、イミプラミン(トフラニール®)、オーラップ(ピモジド®→販売中止)などはQT延長を来しうるのでLQTSと診断されている患者への投与は禁忌である。種類も多く多彩であるため注意が必要である。ADHDなどで投薬を受けているケースは注意深く病歴を取ることが重要である。近年ではアリピプラゾール(エビリファイ®)ブレクスピプラゾール(レキサルティ®)などQT延長作用の小さい薬剤が開発され、選択されるようになっている。これら副作用の少ない薬剤の登場により、ピモジドなどの薬剤は販売中止となっている。
以下にモーズレイ処方ガイドラインの記載をまとめると、
1. QTへの影響の報告なし:アリピプラゾール・ルラシドン・ブレクスピプラゾール
2. 過量投与のみで重度のQTc延長が報告されているか、臨床用量で(<10msec)が観察されている:アセナピン・クロザピン・フルフェナジン・フルペンチキソール・ペルフェナジン・プロクロルペラジン・オランザピン・パリペリドン・リスペリドン・スルピリド
3. 通常臨床用量で平均>10msecのQTc延長が認められるか、特定の状況で心電図のモニタリングが公式に推奨されている:アミスルピリド・クロルプロマジン・ハロペリドール・イロペリドン・レボメプロマジン・メルペロン・クエチアピン・ジプラシドン
4. 通常臨床用量で>20msecの著明なQTc延長が認められる薬剤:抗精神病薬の静注剤すべて・ピモジド・セルチンドール・推奨された用量を超える投薬
5. 作用不明:ロキサピン・トリフルオペラジン・ピポチアジン・ズクロペンチキソール
抗うつ薬
既報によれば、1,497,333人分の心電図結果からシタロプラム、エスシタロプラム、およびアミトリプチリンについて東予量に応じてQT間隔が延長していることが示されている。
(3) 抗生物質
抗生物質の中でQT延長と関わるものと言えば、マクロライド系である。エリスロマイシンやクラリスロマイシン、アジスロマイシンなどは上気道炎や慢性閉塞性呼吸器疾患(COPD)などで処方を受けるケースが多いと思われる。
エリスロマイシンによる分子メカニズムはhERGチャネルをブロックすることで再分極プロセスを延長させていることが示されている。エリスロマイシンを経静脈的に投与した際のQTc変化として、500㎎/分で42ミリ秒、500㎎/20分で33ミリ秒の延長が観察された。細胞実験では10mg/Lでは変化を認めないが、20-5-㎎/Lから再分極過程の延長が観察される。ヒトへ500㎎を15分で静脈注射した際3分後に45㎎/Lになる。
う
更にCYP3A4の酵素阻害の原因となるため、ジソピラミドやキニジンの併用に注意する。
2004年から2011年の間にマクロライドと①トルサード・ド・ポアンツ、②QT延長、③心室性不整脈、④心臓突然死との関連は以下の通り。
全体:①+②は183例、③+④は419例、
クラリスロマイシン:①+②は84例、③+④は162例、
アジスロマイシン:①+②は63例、③+④は140例
エリスロマイシン
最初にエリスロマイシンと不整脈の関連を示された13例のうち12例ではほかにもいくつかのリスク因子を持っていた。全くリスク因子のなかった唯一の例はマクロライド静注を受けた8日齢の乳児であった。
別の報告で、エリスロマイシンとトルサード・ド・ポアンツや心室頻拍との関連が疑われた23例の連続症例では14人が既往に心疾患があり、残り9人のうち5人はエリスロマイシンの静注を受け、2人は他のQT延長の原因となる薬剤を内服し、1人は肝障害を患っていた。結論はエリスロマイシンの静注はQT延長から致死性不整脈の原因となり得ると考えられる。経口エリスロマイシンによる不整脈の報告は、先天性QT延長症候群の患者、次いで抗不整脈薬Ia群であるジソピラミドを内服していた症例、最後に4gのエリスロマイシンを内服した洞性徐脈の患者でラニチジンの常駐の後に症状が現れた。
クラリスロマイシン
それほど症例は多くない。1例目は肺高血圧症、肺性心、低アルブミン血症の患者、2例目はC型肝炎と心不全、透析中の患者、次の4人はいずれもQT延長の原因となる薬剤を併用していたか、先天性QT延長症候群か、低カリウム血症の患者であった。
アジスロマイシン
2013年、米国でマクロライド系が処方された患者数は約5,700万人で、うち90%はアジスロマイシンの処方を受けていた。不整脈を認めた患者は抗不整脈薬第Ia群のジソピラミドを受けていたか、先天性QT延長症候群、うっ血性心不全と診断されていた。
(4) 抗真菌薬
フルコナゾール
フルコナゾールによるQT延長作用についていくつか症例報告がされている。パッチクランプ法を用いた細胞実験によってhERGチャネルの細胞内のトラフィッキングを阻害し、hERGが細胞膜に発現するのを阻害していることが示された。
(5) 抗アレルギー薬
抗アレルギー薬もQT延長の原因となる薬剤の一つである。ガイドラインにはテルフェナジンとアステミゾールが挙げられている。どちらも既に販売中止となっている。アレグラなどの新規抗ヒスタミン剤にQT延長作用の報告はない。
5.QT延長症候群の診断、リスク評価
QT間隔は様々な要因で延長するため、先天性QT延長症候群の診断は症状、心電図所見や様々な負荷心電図、家族歴からスコアリングによって臨床診断となる。

6.心臓の動き(活動電位とは)
心筋細胞の活動電位(action potential、動画1 5’00くらいから、動画2)は、電気的刺激に応じて細胞膜に生じる一過性の膜電位の変化である。活動電位はスパイクやインパルス、あるいは「発火」と呼ばれる。活動電位は、主としてナトリウムイオン(Na+)やカリウムイオン(K+)が、細胞内外の濃度差に従ってイオンチャネルを通じて受動的拡散を起こすことによって起こる。活動電位はさまざまな種類の細胞から生み出されるが、最も広範には神経系において神経細胞同士や、神経細胞から筋肉や腺などの他の体組織に情報を伝達するために使われる。活動電位はすべての細胞で同じというわけではない。心筋は神経に次いで活動電位を発する組織である。
心筋細胞の内と外の間では、電位差が常に存在している。これは細胞内外での金属イオン分布と、これらイオンに対する細胞膜の透過性と選択性に由来する。電荷を持つイオンの分布が細胞内外で異なるため、活性化していない静止状態の細胞では通常、細胞外と比べ細胞内の電位がマイナスとなっている。活動電位とは、この電位差がなんらかの刺激によって一時的に逆転する現象である。
活動電位における負から正への電位の変化に要する時間は短く、数ミリ秒である。どんな細胞の活動電位にも順に脱分極相、再分極相があり、多くの場合過分極相の段階もある。脱分極のプロセスにおいて心筋細胞の興奮、つまり細胞の電位が一時的にプラスからマイナスになる膜電位の一連の反応を通して0mVに近づくことで膜電位が脱分極し閾膜電位(threshold)に達すると活動電位が生じる。心臓のペースメーカー細胞のような細胞では、中間電圧のプラトー相は下降相に先行し活動電位持続時間を数百ミリ秒延長する。活動電位の発生には電位依存性Na+チャネルの存在が不可欠である。心臓の調律を担っている洞房結節の細胞はNa+チャネルの代わりに電位依存性Ca2+チャネルを通してCa2+電流による活動電位を発生させる。
心筋細胞の活動電位による膜電位の変化は以下のプロセスを辿る(図)。
・第0相 Phase0:脱分極相(depolarization)
・第1相 Phase1:スパイク(spike)
・第2相 Phase2:プラトー相(plateau)
・第3相 Phase3:再分極相(repolarization)
・第4相 Phase4:静止電位(resting potential)

心筋細胞の活動電位は神経の活動電位と原則的には同一ですが、0電位付近での再分極の速度が極端に遅いのが特徴的である。活動電位波形の上でこの部分を プラトー相 plateau phase と呼ぶ。心筋活動電位はこのプラトー相を有することが特徴であり、このために活動電位の持続時間が非常に長い。心筋活動電位の各相は、第0相から第4相までの番号で呼ばれる。図に各相の名称を対比して示した。
プラトー相形成のイオン機構は、種々の実験から現在ではほぼ図のように考えられている。Na+流入に対する細胞膜の透過性が急速かつ一過性に上昇することによって細胞内の陽イオン濃度が上がることで活動電位の立ち上り(第0相)が形成される。スパイク形成(第1相)に続いて主としてCa2+に対する透過性がゆっくりと上昇するとともにK+に対する細胞外への透過性が生じ始めることによってプラトー相が形成される。次いでCa2+透過性が減少しはじめK+の細胞外への透過性が上昇することで再分極が起こる。これら金属イオンに対する膜の透過性のもとは、細胞膜に存在するそれぞれのNa+、Ca2+、K+イオンチャネルが担っている。
最初の急速かつ一過性のNa+透過性上昇をもたらす細胞膜のイオン通路を 速いNa+チャネル、これに対してプラトー相形成に関与するものを 遅いCa2+チャネル と呼ぶ。この「遅いCa2+チャネル」はプラトー相形成に関与するとともに、心筋収縮にも極めて重要な役割を果たすと考えられている。このCa2+チャネルは膜電位に依存して活性化されるので、「 電位依存性Ca2+チャネル 」とも呼ばれ、一方、これとは異なるタイプのCa2+チャネルも見出されており、それらと区別するために、 L-型カルシウムチャネルと呼ぶ。
活動電位の発生が細胞内外の陽イオンの濃度勾配を基にしていることは神経の場合と同様であり、Na+とK+濃度勾配の維持にはNa+ポンプが関与している点も神経と同じである。Ca2+の濃度勾配(細胞外が高く、細胞内はきわめて低い)維持にはCa2+ポンプやNa+/Ca2+交換機構が関与すると考えられている。

7.心筋細胞レベルで起こっていること
QT延長や致死性不整脈の発生は心筋細胞にあるイオンチャネル機能に関係する遺伝子異常によって起こっている。何故遺伝子変異があるとQT時間が延長し、トルサード・ド・ポアンツのような不整脈が発生するのか?多くの場合、心室筋細胞の外向き電流(LQT1:IKs, LQT2: IKr)が減少(loss of function)するか、または内向き電流(LQT3: late INa)が増加(gain of function)することにより活動電位持続時間(APD)が延長し、心電図上のQT時間の延長を呈する。さらに活動電位のプラトー相付近の時間が長くなることから内向きCaチャネルが再活性化し、早期後脱分極(EAD)を生じると不整脈につながると考えられる。
KCN〇やSCN〇とはK-ChaNnelやSodium-ChaNnelから取られた略語である。KチャネルはPCNでは?と思うかもしれないが、会話では「ケーチャネル」という言葉を使うためだろう。Naチャネルは「ナトリウムチャネル」や「ソディウムチャネル」と呼称している。
個々のチャネルの機能:
(1) LQT1, KCNQ1
QT延長症候群のLQT1の原因遺伝子はKCNQ1である。このまとめが分かりやすい。6開幕貫通型タンパク質の電位依存性カリウムチャネルであるKv7.1をコードし、主に心筋活動電位の再分極を担う。KCNQ1サブユニットが4つ集まり1つのチャネルを形成する。また機能修飾サブユニットであるKCNE1が結合し、チャネルの開孔(=ゲーティング)の調節に関わっている。
心筋にある3つタイプのKチャネルのうち「遅い」遅延整流性カリウム電流IKsを担っている。NaチャネルやCaチャネルによって脱分極、つまり心筋細胞内で負電荷に分極している電位を陽イオンが流入することによって、0mV 近くまで電位まで分極から脱した際に、Kチャネルから陽イオンが外に排出され始めて再び細胞内電位を負電荷に分極させること、「再分極」プロセスに関わっている。Kチャネルは閉状態から中間状態を経て開状態となる。細胞内膜電位が負の分極から脱すると電位センサーによってチャネル構造が変化しポアが開く。このチャネルは電位の変化によって開孔まで時間がかかり、閉じるのも時間がかかる。
サブユニットの分子構造は最初の4つの膜貫通ドメインS1-S4が電位センサーで、残る2つのS5,S6がポア(孔)を形成する。このポアの部分は4つのサブユニットは中央に寄り集まって形成され、電位センサーのドメインは周辺で独立して機能している。電位センサーのS4の正電荷アミノ酸(アルギニン等)によってまく電位を感知し、S4とS5をつなぐリンカーを介してポアドメインが開閉すると考えられている。
KCNQ1に遺伝子変異が生じると機能喪失(Loss-of-function)を起こしチャネルが開きにくくなるため電流が減少するので、再分極つまり心臓細胞内の電位が腑に戻りにくくなり、QT延長の原因となる。
(2) LQT2, KCNH2
QT延長症候群のLQT2の原因遺伝子はKCNH2である。このまとめがよい。電位依存性カリウムチャネルであるKv11.1をコードし、主に心筋活動電位の再分極を担う。hERG (”ハーグ”)と呼ぶ人もいる。
KCNH2がコードするKv11.1は6回膜貫通型タンパク質(S1からS6)であり、電位センサーであるS1からS4と孔(pore)のS5,6から成る。K+を細胞内へ流入させる。
心筋にある3つタイプのKチャネルのうち「速い」遅延整流性カリウム電流IKrを担っている。NaチャネルやCaチャネルによって脱分極、つまり心筋細胞内で負電荷に分極している電位を陽イオンが流入することによって0mV電位まで戻った後、Kチャネルは再び細胞内電位を負電荷に分極させること、再分極プロセスに関わっている。
HERGチャネルには3つの形態がある。閉孔、開孔、不活化である。これらの3つの状態間の遷移は、電圧に依存する。活性化は細胞内ゲートの開放を含み、一方、不活性化は透過経路の細胞外末端でのゲートの開放を伴う。このチャネルは電位の変化によって開孔まで時間が早く、閉じるのも早い。
実験でパッチクランプ法を物し給う尊敬すべき先輩たちはこちらの方を好んで使っている。たぶん理由は名前の由来がユニークだから。この遺伝子に変異が生じたショウジョウバエをエーテルで麻酔すると、ダンスするように脚を震えさせたことから、カリフォルニア州ウェスト・ハリウッドのナイトクラブ「ウィスキー・ア・ゴーゴー」において当時人気であったダンスにちなんでether-a-go-go遺伝子と命名された。
(3) LQT3, SCN5A
QT延長症候群のLQT3の原因遺伝子はSCN5Aである。このまとめが分かりやすくておススメ。Nav1.5をコードする電位依存性膜貫通性のタンパク質である。4つの反復する膜貫通ドメインを持ち、それぞれのドメインにS1からS6のセクションを持つ、巨大なたんぱく質である。主に心筋活動電位第0相の脱分極過程を担う。Kチャネルとは異なり、1つのタンパク質で1つのαサブユニットとなる。4つの機能修飾βサブユニットであるSCN1B, SCN2B, SCN3B, SCN4Bと結合し、チャネルの開孔(=ゲーティング)の調節に関わっている。
Nav1.5は活性化したのち、不活性化が極めて速い。SCN5Aの機能獲得型の遺伝子変異によって遅延Na電流が残存し、不活性化が不十分になってしまうこと、また再分極過程で部分的なウィンドウ電流が発生することで活動電位幅が延長することでQT延長をきたす。このようなNaイオンの持続的流入に伴って起こる細胞内濃度の上昇によって、細胞内Caイオンの過負荷につながり、心筋収縮・弛緩、酸素消費の過程に負の影響を及ぼす可能性がある。
LQT3に対するβ遮断薬による薬物療法は前臨床研究において有害である可能性が示されていたが、その後の臨床研究を通して有益であることが示されている。直接的な薬理学的アプローチはL型Na電流をブロックするメキシレチン、フレカイニド、ラノラジンである。
同じSCN5Aの遺伝子変異でも機能喪失型の場合は、Na電流の活性化の遅延あるいは早い不活性化が生じ、ブルガダ症候群の原因となる。
8.先天性QT延長症候群の治療
(1) 生活指導
遺伝子型によって生活上注意すべき点は異なる。運動は、LQT1とLQT2において制限されるべきである。またLQT2については加えて音刺激のような情動ストレスに注意すべきである。LQT3は睡眠や安静時に発作が多く、生活上の注意で発作を予防することは難しい。全ての型について共通して言えることはQT延長作用のある薬剤(BOX1)は服用しないように、他の医療機関や薬局を訪れる際には必ず本疾患名を告げるようにする。
(2) 運動制限
米国ではBethesdaのガイドラインに基づき、QT延長症候群患者はスポーツ競技に参加できない。条件は(1)症状、(2)470ms(男性)/489ms(女性)を超えるQTc間隔、(3)ICD植込み患者、である。欧州では欧州心臓病学会ガイドラインで440ms(男性)/460ms(女性)を超えるQTcのカットオフのみに基づきスポーツ競技参加を失格としている。
LQT1以外の低リスク例ではQT間隔が正常か境界域である。無症状かつ突然死の家族歴もない例では全ての検査で評価し、また運動中もAEDの準備をし、心肺蘇生の講習を受けたスタッフの監視下であればある程度のスポーツ競技を行うことは許可される、という考え方がある。
ある報告で、353人のLQT1-3患者のうち、130人(37%)は診断後もスポーツ競技に参加していた。うち20人はICD植込み術後である。そのうち9歳男児1人がスポーツ中に心イベントを起こした。LQT1でQTc>500ms、心停止の病歴あり。試合前のウォーミングアップ時に発生。
(3) 妊娠・出産
もっとも有名な論文で、QT延長症候群の女性が妊娠・出産の際に経験した心イベントに関する調査がある。1980年から2003年の間に登録された391人について産褥期には心イベントがLQT2を除いて低下し、その後は元のリスクに戻っていた。LQT2女性についてもβ遮断薬の内服によってリスクが低下していた。
基本的にLQT1妊婦に心イベントのリスクは高くはないが、β遮断薬の継続的な内服はリスクを下げる可能性がある。115人の妊娠したLQT1保因者のうち3人の心イベントが発生した(うち2人は心停止)。うち有症状27人中11%ということになるが、β遮断薬内服例では発症者はいなかった。変異保持者と対照者で流産の発生率は内服の有無で差はなかったが、帝王切開は保因者群で多かった(27% vs 14%)。Β遮断薬内服と胎児への影響には差がなかった。胎児機能不全の乳児は全てA341V変異保持者であった(10人全員)。
(4) 薬物治療
薬剤の中心はβ遮断薬であるが、遺伝子型によって治療方針は変わる。β遮断薬はQTcを短縮させることはないが、心イベントの発生リスクを軽減させる。LQT1では74%、LQT2では63%のリスク低下がみられた。LQT3では女性のみ83%のリスク低減を認めたが、男性では認めていない。
β1非選択性β遮断薬(プロプラノロールやナドロール)の方がβ1選択性β遮断薬(アテノロールやメトプロロール)より有効性が高い。LQT2にはナドロールが勧められている。
LQT3例では,遅発性 INa遮断薬であるメキシレチンがQT間隔を短縮し,心イベントの予防に有効と考えられている。LQT7(Andersen-Tawil症候群)例では,CPVTと同様にフレカイニドが有効である。
Ca拮抗薬はトルサード・ド・ポアンツの発生抑制に寄与すると考えられているが、エビデンスとしては確立していない。β遮断薬のみでは再発を完全に抑制できない例に併用で処方される場合が多い。なおLQT8では有効例が報
告されている。
(5) 非薬物治療
心室細動や心停止の既往を有する患者は植込み型除細動器(implantable 、cardioverter defibrillator; ICD)のクラスI適応である。心室細動や心停止がなくても、①トルサード・ド・ポアンツまたは失神の既往、②突然死の家族歴、③β遮断薬に対する治療抵抗性のうち2項目以上を認める場合はクラスIIa、1項目を認める場合はクラスIIbのICD適応となる。ただし、LQT3では①と②のいずれかを認めればクラスIIaのICD適応となる。
9.先天性QT延長症候群の原因遺伝子
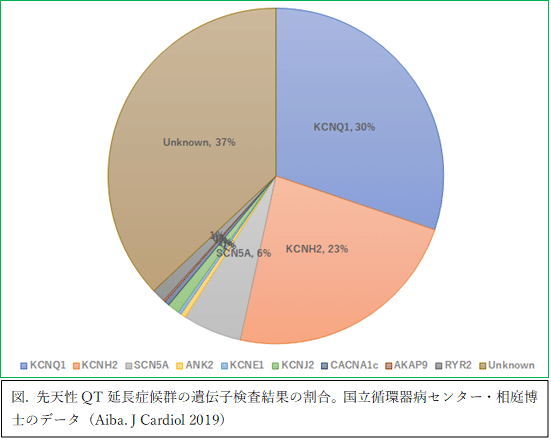
LQTSは主に心筋細胞の膜表面に発現するイオンチャネルをコードする遺伝子の変異によって引き起こされる。過去10年間で17の遺伝子が同定され、先天性QT延長症候群はのうち常染色体優性遺伝形式のものはRomano-Ward症候群として順番にLQT1, LQT2, LQT3・・・LQT16と命名されている。常染色体劣性遺伝形式のものはJervell-Lange-Nielsen症候群として原因遺伝子はJLN1, JLN2と命名されている。実際に遺伝子と疾患の関連を強く示す遺伝子は限られており、近年では原因遺伝子を整理する傾向がみられる。その中で特に3つの遺伝子、KCNQ1、KCNH2、SCN5Aの原因となる変異は、QT延長症候群の臨床診断を受けた患者の約60〜70%で検出される。他の遺伝子のうちAKAP9、KCNE1、KCNE2、KCNJ2、KCNJ5、SCN4B、SNTA1については今後も検討が必要と考えられる。CALM1、CALM2、CALM3、TRDNはそれぞれ患者の1%未満であり新生児房室ブロックを伴うLQTSを示すことが判明した。またティモシー症候群(CACNA1C)、アンデルセン-タウィル症候群(KCNJ2)はQTcが延長する稀少な多系統症候群に含まれる。
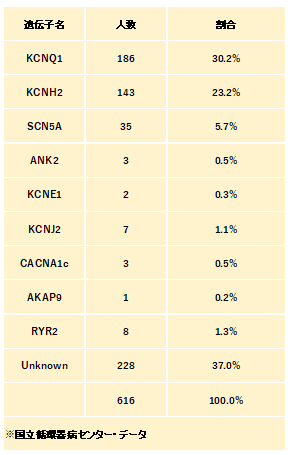
10.個々の遺伝子型
(1) LQT1
LQT1の原因遺伝子はKCNQ1という遺伝子であり、日本人のLQTSの30.2%が遺伝子の変異が同定されており、最も頻度が高い。KCNQ1(NM_000218.3)は電圧に依存して開口するカリウムチャネルのうちサブファミリーQメンバー1(potassium voltage-gated channel subfamily Q member 1)である。
補正QT(corrected QT; QTc)が530ミリ秒を超える患者は500ミリ秒以下の患者に比べて3.25倍心イベントが多い。初回新イベント発生年齢も全ての遺伝子の中で最も低い。
LQT1ではカリウムイオンが通過する孔(pore; 穴のような通り道のこと)の領域を含む膜貫通領域での変異で新イベントの発生率が高い。
男性の場合変異部位による心イベントの発生率に違いがないが、女性の場合は孔領域での変異の心イベント発生率が他の変異に比べて高いことが知られている。
LQT1では運動制限が必須であり、「競技レベル」の運動、特に競泳、潜水は禁止とする。薬物治療としてはβ遮断薬が最も有効で、74%の新イベントリスク低下効果を認める。特にβ1非選択性のβ遮断薬の有効性が高い 。
(2) LQT2
LQT2の原因遺伝子はKCNH2という遺伝子であり、日本人のLQTSの23.2%を占めている。KCNH2(NM_000238.4)は電圧に依存して開口するカリウムチャネルのうちサブファミリーHメンバー2(potassium voltage-gated channel subfamily H member 2)である。
補正QT(corrected QT; QTc)が530ミリ秒を超える患者は460ミリ秒以下の患者に比べて3.33倍心イベントが多い。
LQT2ではカリウムイオンが通過する孔(pore; 穴のような通り道のこと)の領域の変異で心イベントの発生率が高い。
心イベントの発生に男女差はない。
LQT2では情動ストレス(恐怖や驚愕)、音刺激(目覚まし時計など)による覚醒時などに心イベントが起こることが多く、運動制限とともに音刺激を避けるなどの生活指導も重要である。薬物治療としてはβ遮断薬が最も有効で、63%の心イベントリスク低下効果を認めるが、他の抗不整脈薬(メキシレチン、ベラパミル)の併用による血清カリウム値の上昇も有効である。
(3) LQT3
LQT3の原因遺伝子はSCN5Aという遺伝子であり、日本人のLQTSの5.7%を占めている。SCN5A(NM_000335.5)はナトリウムチャネルのサブユニット5(sodium voltage-gated channel alpha subunit 5)である。これはブルガダ症候群のリスクをあげる遺伝子でもある。
補正QT(corrected QT; QTc)が500ミリ秒を超える患者は心イベントが多い。
LQT3ではナトリウムイオンが通過する孔(pore; 穴のような通り道のこと)の領域の変異で心イベントの発生率が高い。
心イベントの発生に男女差はない。失神や致死性イベントを合わせた「総イベント数」の発生率はLQT1、LQT2に比べてLQT3では少ないが、総イベント数のうち致死性イベントの割合は高いと考えられている。
LQT3では睡眠中や安静時に心イベントが多く、生活指導による発作予防には限界がある。薬物治療としてはメキシレチンが有効であるが、女性ではβ遮断薬が有効であると報告されている。
(4) LQT4
LQT4の原因遺伝子はANK2、Ankyrin2である。この遺伝子は細胞膜の内側で膜タンパク質を基盤をなすスペクトリン-アクチン細胞骨格にリンクするアンキリンファミリーのタンパク質のメンバーをコードしている。日本人での頻度は極めてまれで0.5%である。連鎖解析を通して同定された。
LQTSの他、洞機能不全やカテコラミン誘発多形性心室頻拍(CPVT)の様な心電図変化を生じる可能性がある。そのため、ANK2変異に伴って生じる一連の不整脈所見を合わせて「アンキリン症候群」と呼ぶこともある。
(5) LQT5
LQT5の原因遺伝子は電位依存性カリウムチャネル・サブファミリーE制御サブユニット1(KCNE1, potassium voltage-gated channel subfamily E regulatory subunit 1)である。日本人での頻度は極めてまれで0.3%である。Romano-Ward症候群とJervell-Lange-Nielsen症候群どちらの原因遺伝子である。
(6) LQT6
LQT6の原因遺伝子は電位依存性カリウムチャネル・サブファミリーE制御サブユニット2(KCNE2)である。日本人ではこれまで変異は認めていない。KCNH2と構造的に類似していることが指摘されている。
(7) LQT7
LQT7の原因遺伝子はKCNJ2、カリウム内向き整流チャネルサブファミリーJメンバー2 (potassium inwardly rectifying channel subfamily J member 2)をコードしている。頻度は1.1%であり、QT短縮症候群の原因遺伝子でもある。この遺伝子によってコードされるタンパク質は、内在性膜タンパク質および内向き整流型カリウムチャネルである。カリウムが細胞からではなく細胞に流入する傾向が強いコード化されたタンパク質は、おそらく活動電位波形と新規細胞の興奮性の関与している。もともとこの遺伝子の変異は、発作性周期性四肢麻痺、形態異常(広い額と眉間部、耳介低位、両眼解離、下顎低形成、合指症)を伴うアンデルセン・タウィル症候群の心症状として報告された。KCNJ2変異保持者には全身症状は軽微で心症状のみ呈する例が存在する。心電図ではU波が顕著であり、QU感覚が延長している。
アンデルセン・タウィル症候群ではフレカイニドが有効である。
(8) LQT8
LQT8の原因遺伝子は電位依存性カルシウムチャネルサブユニットα1C(CACNA1C; calcium voltage-gated channel subunit alpha1 C)である。日本人での頻度は非常にまれで0.5%である。LQTSだけでなく、ブルガダ症候群の関連遺伝子、あるいはティモシー症候群の原因遺伝子でもある。
βアドレナリン作動性アゴニストによってこの電位依存性カルシウムチャネルを刺激するメカニズムが確立されている。ベラパミルによってティモシー症候群でみられる心室頻拍を抑制するという報告がある。
※ティモシー症候群(TS; OMIM 601005)は、致命的な不整脈、指や足の指の水かき、先天性心疾患、免疫不全、断続的な低血糖、認知異常、自閉症などの多臓器機能障害を特徴としている。
(9) LQT9
LQT9の原因遺伝子はカベオリン3(CAV3; caveolin-3)である。日本人では変異を認めていない。乳幼児突然死症候群の原因遺伝子の候補でもある。50人の黒人乳幼児のうち3つのミスセンス変異を認めているが、83人の白人には認めていない。
カベオリン3の遺伝子変異の機能獲得によってナトリウム電流の増加が実験上認められている。同様の所見が乳幼児突然死症候群でも見られている。
(10) LQT10
LQT10の原因遺伝子は電位依存性ナトリウムチャネルβサブユニット4(SCN4B; sodium voltage-gated channel beta subunit 4)である。日本人では遺伝子変異を認めていない。東アジア人種で家族性心房細動の原因遺伝子でもある。
SCN2Aと共有結合をし、チャネル特性を変更する機能を持つ。
(11) LQT11
LQT11の原因遺伝子はAキナーゼ・アンカータンパク質9(AKAP9;A-kinase anchor protein 9)、別名Yotiaoである。日本人の頻度は極めてまれで0.2%である。
AKAP9はKCNQ1のアルファサブユニット、KCNE1の調節サブユニット(KCNE1; 176261)、およびKCNQ1C末端のロイシンジッパーモチーフに結合してPKAに結合している。AKAP9の調節異常が突然死との関連を示されている。
(12) LQT12
LQT12の原因遺伝子はシントロフィンα1(SNTA1; Syntrophin 1α)である。日本人では変異を認めていない。シントロフィンは、ジストロフィン関連タンパク質複合体の成分である細胞質表在性膜足場タンパク質である。この遺伝子はシントロフィン遺伝子ファミリーのメンバーであり、心臓組織に見られる最も一般的なシントロフィンアイソフォームをコードしている。このシントロフィンタンパク質のN末端PDZドメインは、心臓のナトリウムチャネルNav1.5の孔形成アルファサブユニット(SCN5A)のC末端と相互作用する。
(13) LQT13
LQT13の原因遺伝子はGタンパク質感受性カリウム内向き整流チャネルサブファミリーJメンバー5(KCNJ5 potassium inwardly rectifying channel subfamily J member 5)である。日本人には変異は認めていないが、中国人家系で示された原因遺伝子である。家族性高アルドステロン症でも認められた原因遺伝子でもある。
(14) LQT14
LQT14の原因遺伝子はカルモデュリン1(CALM1)である。EFハンドカルシウム結合タンパク質である。カテコラミン誘導性多形性心室頻拍の原因遺伝子の一つである。日本人では変異を認めていない。
(15) LQT15
LQT15の原因遺伝子はカルモデュリン2(CALM2)である。EFハンドカルシウム結合タンパク質である。カテコラミン誘導性多形性心室頻拍の原因遺伝子の一つである。日本人において、LQTSとCPVTの両方の原因になり得るという根拠がある。
(16) LQT16
LQT16の原因遺伝子はカルモデュリン3(CALM3)である。EFハンドカルシウム結合タンパク質である。カテコラミン誘導性多形性心室頻拍の原因遺伝子の一つである。日本人では変異を認めていない。
12.結果の解釈について
遺伝子検査において原因遺伝子の中に変異が同定されてもその変異が疾患発症の原因となりうるかどうか結果の解釈をする必要がある。疾患の有無にかかわらず稀少変異は存在しうる。病的変異が確認されている家系内においても頻繁に認められる浸透度の変動や散発的な遺伝子型陰性の症例の存在によって、多くの稀少変異が一般集団内で予想外に高い頻度で認められていることから、臨床的に関連する遺伝的変異の中に病原性のスペクトルが存在すると考えられている。単独で疾患の原因となるには不十分な低および中程度の浸透性変異が存在することから、遺伝性不整脈の病因は遺伝的要因のみならず、臨床的・環境的要因も重要であることが共通見解となっている。
変異は病的、良性、病的意義の不明な変異(VUS)に分類される。因果関係に十分な証拠がある場合、あるいは変異が疾患を引き起こしていないことを裏付ける根拠が明確な場合は、それぞれ病的変異・良性変異と診断できる。ところが因果関係が不十分である場合、あるいは矛盾している場合はVUSとして分類される。2015年、米国臨床遺伝・ゲノム学会(ACMG)は変異解釈に関する共同声明を発表しされた指針が広く採用されている。ClinVarやHGMDなどの変異情報の公開リポジトリからのデータは解釈に役立つ。健常人の遺伝情報についてはgnomAD v3.1や東北メガバンクの日本人多層オミックス参照パネル(jMorp)において確認できる。日々情報が更新されており、以前の分類の根拠となったデータに新しいデータを加えて再評価することが重要である。遺伝子が特定の表現型と強い関連を示すかという点は更に重要である。ClinGenのサイトにおいて遺伝子型と表現型をリンクの臨床および実験的証拠を評価する必要されている。
13.複雑な遺伝病と多遺伝リスクスコア
ヒトの形質は多数の遺伝子と環境要因の組み合わせで発現する。メンデル遺伝形質は、ある1つの座位の特定の遺伝型の存在だけでその形質の発現に必要かつ十分となる、遺伝性疾患の中でも最も単純な形質である。遺伝性疾患・形質の多くはメンデル遺伝形質ではなく複数の座位の遺伝子群によって発現が制御される非メンデル型遺伝形式である。これまで遺伝性不整脈はこのメンデル遺伝形質の文脈の中で説明されてきた。遺伝子解析の結果、家系解析においても浸透率の低い変異がみられ発症に寄与しているものの変異のみでは発症しないものも存在する。近年の遺伝子解析研究の結果、そのため浸透率が低い遺伝性不整脈も見られる。例えば、Brugada症候群はSCN5A以外にも多くの遺伝子が候補遺伝子として同定されているが、臨床的な意義は現在も論争となっている。遺伝子検査で変異を認めないBrugada症候群などの例では全体として主要なドライバーが寄与している可能性が考えられている。
ポリジーン器質は、冠動脈疾患や心房細動などのように、多くの遺伝的要因および環境的・後天的な要因によって引き起こされる。ゲノムワイド関連研究(GWAS)を通して、一つ一つの疾患リスクの効果量は小さいが、数千の一塩基多型(SNP)を特定されている。多遺伝子リスクスコア(PRS)は、これらの多型の相加効果であり、疾患を発症するリスクを予測するために使用できる。PRSが冠動脈疾患の患者の管理において重要な予後的価値を提供する可能性を示した。何百万もの多型を組み合わせることにより、PRSが計算され、単一の従来の危険因子よりも冠動脈疾患のより良い予測が示された。まだ明確な臨床応用には至っていないが、PRSを将来的には臨床情報と組み合わせて使用することで、複雑な疾患や疾患発症リスクが高い人の予測性能を上げ、健康増進に寄与する可能性がある。
14.遺伝カウンセリング
遺伝子検査の特殊性として、ある個人での検査結果は生涯変更がないこと、1つの変異は次世代へ50%の確率で受け継がれることである。ここから健常人でも将来重篤な疾患を発症する可能性を知りえることになり、また血縁者が病的変異保因者であることから発症リスクを知り得ることとなる。そのため遺伝診療においては各段階で遺伝カウンセリングが不可欠である。がんゲノム診療体制には遺伝カウンセラーが必ず含まれ、遺伝子検査が患者のみならず健康な家族へも及ぼし得る影響についての情報提供や病的変異陽性者やその家族への心理的サポートに欠かせない存在となっている。
遺伝診療は明確に診断されている発端者について原因遺伝子の変異同定を実施したのち、カスケード検査として同じ変異について無症候性のリスクのある親族に提供するという2段階のプロセスが一般的な流れである。遺伝子検査を受ける人を厳密に選択し、変異について正確な解釈をすること、この二点が患者とその家族にとって最良の結果が達成されるために重要である。被検者の選択基準として、特徴的な表現型を示していること、若年発症、重篤さ、濃厚な家族歴などは遺伝的な影響を強く示唆する。臨床診断が不明確なまま検査を進めても同定された遺伝的変異の解釈が非常に困難となる。遺伝性不整脈の場合、誤分類によって重大な影響を及ぼす可能性があるため検査の事前確率が低い場合は避けることが望ましい。家族歴の取得は遺伝診療の第一歩と言える。遺伝性不整脈は一般的に常染色体優性(AD)の遺伝形質を取る。心臓突然死や失神は明確な症状である反面、様々な要因でも起こりうるため、死後の情報を含めて詳細な問診が必須である。その中で専門的なスキルを持つ遺伝カウンセラーは遺伝リスクについて良好なコミュニケーション関係を構築し、遺伝子検査やサーベイランスの頻度の提案を通して、より良い心理社会的適応に導く役割を果たしている。
この記事が気に入ったらサポートをしてみませんか?