AutoDockToolsを用いてAutoDock4を実行(備忘録)

ファイルの準備
AutoDockToolsを使う前にディレクトリを作成して、その中に、protein.pdbとligand.mol2を入れておく。
(ドッキングシミュレーションして、リガンドの初期構造が結構大事そうだから、エネルギー最小化させたリガンドのmol2ファイルの作成をしておきたい。WindowsならChem 3D)
AutoGridの準備
XQuartzを開き、xtermのターミナルで、以下のように打ち、AutoDockToolsを開く。
$ adt開いたら、以下のように実行する。
protein.pdb→protein.pdbqtへの変換
File > Read Molecule > # protein.pdbを開く
Edit > Delete Water #水を取り除く
Edit > Hydrogens > Add > そのままYes #水素を付加
Edit > Charges > Add Kollman Charges #タンパク質各原子に電荷を割り当てる
Edit > Atoms >Assign AD4 types #AD4タイプへの割り当て
Grid > Macromolecule > choose > proteinを選択> Select Molecule > protein.pdbqt 保存
ligand.mol2→ligand.pdbqtへの変換
Ligand > Input > open > ligandを選択 > Select Molecule
Ligand > Input > choose > ligand
Ligand > Torsion True > Detect root
Ligand > Torsion True > choose torsion
Ligand > Output > Save as PDBQT
タンパク質(受容体)の一部残基を動かしたいとき
Select > Select From String > Residueのリストへ ”ASN89, GLY157”と入力
> Add > Dismiss (多すぎると実行できなくなる)
Flexible Residues > Input > Open Macromolecule…でpdbqtファイルを開く > Choose Macromoleculeで選択
Flexible Residues > Choose Torsions …
Flexible Residues > Output > Save Flexible PDBQT…とSave Rigid PDBQTで保存
GPFファイルの生成
Grid > Macromolecule > choose > rigid.pdbqtを選択> Select Molecule
Grid > Set map types > Directly >そのままAccept
Grid > Grid Box… > #グリッドサイズを決めたら、File > Close saving current
Grid > Output > Save GPF #grid.gpfを保存
AutoGridを実行
Run > AutoGrid #入力して > Launch
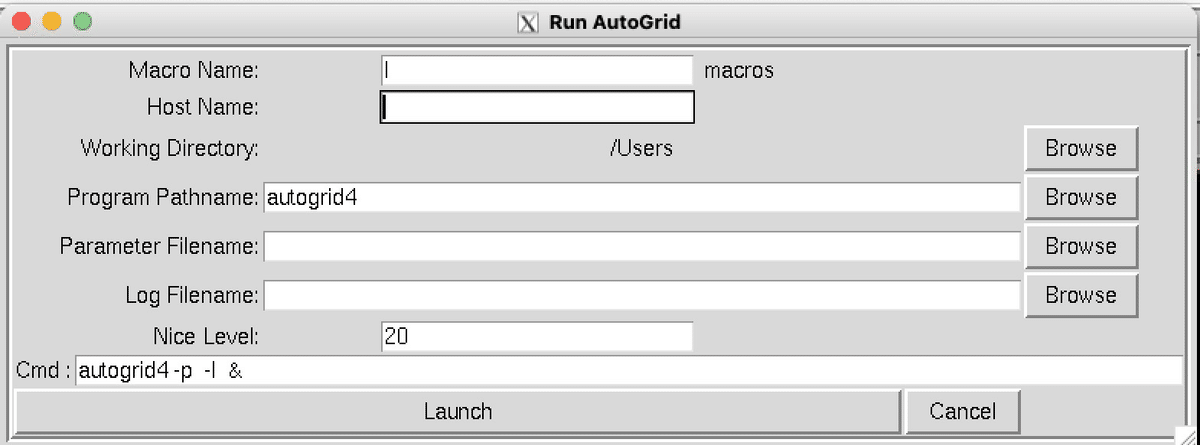
Macro Name, Host Nameはすでに入っているはずです。
Working Directory:pdbqtファイルなどを保存しているところ
Program Pathname:autogrid4があるところ(autodockをダウンロードした場所)
Parameter Filename:gpfファイルをBrowseで探す。
Log Filename:パスさえ通っていれば、gpfファイルを入れただけで自動的に入力される。
Nice Level:よくわからない。そのまま
AutoGridの実行が成功しているかどうか確かめる。
$ less grid.glg最後に"Successful Completion."と書いてあればOK。
Dockingの準備
DPFファイルの作成
# ADT上でプロテインのみを表示させる(リガンドを左のボタン押して消す(Deleteではない))
Docking > Macromolecule >Set Rigid Filename >proteinを選択
※Flexibleも必要があれば選択する
Docking > Ligand > choose > ligandを選択 > そのままAccept
Docking > Search Parameter > Genetic Algorithm > 10→100 > Accept
Docking > Output > Lamarckian > #dock .dpfを保存
AutoDockの実行
Run > AutoDock #入力して > Launch
AutoGridと同じような画面が出てくる。
違うところ↓↓
Program Pathname:autodock4があるところ(autodockをダウンロードした場所)
Parameter Filename:dpfファイルをBrowseで探す。
結果を解析
結果は、生成したdlgファイル内にある。
$ less dock.dlgこのような結果がどこかしらにある。。

解析には、一回ADTを閉じて、もう一度開く。
Analyze > Docking > Open #dlgファイルを開く。
Analyze > Macromolecule > Open #protein.pdbqt
Analyze > Conformations #再生する # &; > show infoとすると、構造と結合自由エネルギーを同時に見ることができる。
Analyze > Clusterings > #グラフが出力 > Edit > Write #保存
参考にした動画
https://youtu.be/ZVKKsK5DsCY?si=LnNE3cXH5_F5rFga