
AlphaFoldの衝撃
DNAには生命の情報が格納されており、RNAに転写され、タンパク質に翻訳されることで機能を発揮する。これは、生物学のセントラルドグマ(中心教義)と呼ばれ、生物学の根幹をなしている。
このうち、20年ほど前から生物のDNA(ゲノム)を解読することができるようになり、生物の設計図が分かってきた。各生物がどの種類のRNAをどの程度転写しているか(トランスクリプトーム)も調べることができ、最近では一つ一つの細胞のRNA発現量を定量する研究(シングルセルトランスクリプトーム)が流行っている。
しかし、多くの生物学的機能を担うのはタンパク質であり、その機能を知るためには構造情報が重要であるが、構造は1種類ずつ実験的に決定することしかできず、網羅的に構造決定すること(プロテオームの構造決定)はできなかった。
生物学はこの60年ほどでセントラルドグマの入り口(DNA)についての理解は深まったが、出口(タンパク質)の理解が圧倒的に追い付いていなかった。(タンパク質のデータバンクに登録されている構造数が約19万に対し、タンパク質をコードする配列として登録されているDNA配列は2億以上)
そんな中、今回紹介する論文であるAlfaFold2は配列からのタンパク質構造予測で実験による構造決定と同程度の高精度を出し、タンパク質科学を新しい時代へと導いている。
Jumper, John, et al. "Highly accurate protein structure prediction with AlphaFold." Nature 596.7873 (2021): 583-589.
DOI: https://doi.org/10.1038/s41586-021-03819-2
図1Aでは、AlphaFoldがCASP14というタンパク質の配列から構造を予測するコンテストで圧勝したこと、図1B,C,Dでは、非常に高精度に予測できた構造例が、図1Eではモデルの概要図が示されている。

図2Aでは、PDBに登録された新規構造について、タンパク骨格のずれが中央値1.46Åという高い精度で構造予測できること、図2Cでは、モデルが予測精度が高いと思っている構造の精度が実際に高いという相関があることが示されている。これは、予測構造の妥当性を評価する際に重要な性質となる。

AlphaFoldのネットワークは構造を調べたいアミノ酸配列と、それと相同なアライメントされたアミノ酸配列を入力として、タンパク質の構造を出力する。ネットワークは2つのメインのパーツからなる。
一方はEvoformerと呼ばれるもので、自然言語処理の分野で有名なattention機構等を用いて多数のアライメントと残基ペアの関係を出力する。この処理によって、配列間やアミノ酸残基間の空間的進化的関係性が表現されているそうだ。
その後は3次元構造を決定するための回転や移動を担うネットワークが続く。モデルの概要図でも示されているように、出力されたロスに基づき反復的に構造の精緻化を行っていくことが最終的な精度の上昇に大きく貢献しているそうだ。(これはコンピュータービジョンの分野で発展した技術)
残基ペアの関係性を表現する際には、残基同士を結ぶ3次元のグラフを考え、3つのノードの三角形を単位として用い、位置関係を改善していく(図3C)。これも成功要因の一つとなっているようだ。
これは、将棋AIが駒の3つ組の位置関係で盤面を表現することで強くなったことを連想させる。DeepMindは過去にAlphaZeroを開発した際に将棋AIとの対戦も行っており、もしかしたらそこから着想を得た可能性もあるのだろうか?

AlphaFoldは過去のツールに比べて大幅に信頼性の高い構造情報が得られ、将来的には日常的に使われるツールとなることが予想される。
ただし、多くのツールと同様にブラックボックスとして使用するのではなく、出力された構造が本当に妥当なものであるのかを確認しながら使用することが大切だそう。
同時期に出版されたヒトプロテオームをAlphaFoldを用いて構造予測したという以下の論文では、現在までに実験的に構造決定されたヒトタンパク質が約17%であるのに対し、36%のタンパク質を非常に高い精度で構造予測し、58%のタンパク質は信頼できる精度で構造予測できたことが示されている。
Tunyasuvunakool, Kathryn, et al. "Highly accurate protein structure prediction for the human proteome." Nature 596.7873 (2021): 590-596.
DOI: https://doi.org/10.1038/s41586-021-03828-1
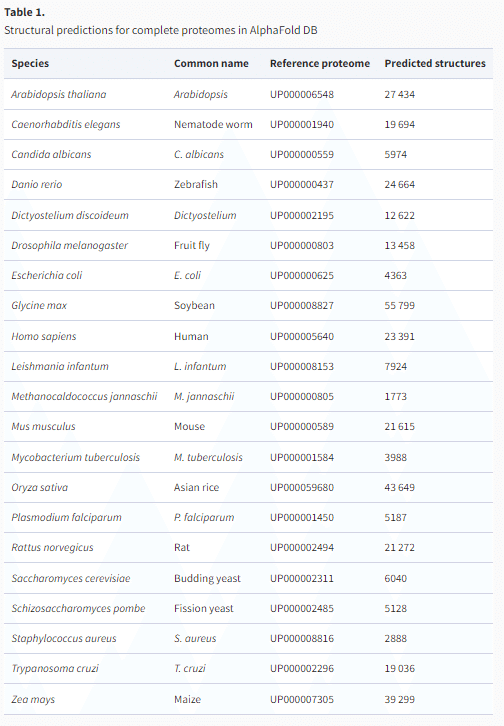
また、現在では以下の表で示されるように大腸菌、マウス、線虫、ゼブラフィッシュなど他の様々なモデル生物のプロテオームも構造予測がなされており、分子生物学の研究を行っている多くの人にとって、注目するタンパク質の構造が手に入る状況となっている。
上記のヒトプロテオーム論文の後半でも行われているが、今後は一人一人の研究者が予測された構造が妥当なものであるか判断し、その生物学的解釈を行う必要があり、構造生物学の素養を持つ重要性が増していくだろう。
AlphaFoldはタンパク質単体の構造予測プログラムであるが、ソースコードがオープンにされたことでタンパク質複合体構造予測、アミノ酸変異がタンパク質の構造に与える影響の予測、タンパク質のde novoデザインへの応用など様々な研究が展開されており、今まで不可能だったり考えもしなかったりしたことが可能になりつつある。
使われている技術としては他の分野で提唱されているものを主に応用しているようだが、図4で示されているように、ここまで徹底的に多数のテクニックを組み合わせて実際に圧倒的なパフォーマンスを出すまでエンジニアリングするのは、やはりDeepMindすごいなあと感じた。

参考文献
タンパク質の構造を予測する論文を紹介している。実験による構造決定と同程度の高精度を出し、タンパク質科学を新しい時代へと導いている。将棋AIが駒の3つ組の位置関係で盤面を表現することで強くなったことを連想させる。
サムネイル画像の出典:https://www.nature.com/articles/s41586-021-03819-2